![]()
|
|
Neurobiologija demencije: Uvod u Alzheimerovu
i druge neurodegenerativne bolesti moždane kore
doc. dr.sci. Goran Šimić, dr. med.
Zavod za neuroznanost Hrvatskog instituta za istraživanje mozga
Medicinskog fakulteta Sveučilišta u Zagrebu
Smrt nije izvan mene. Ona je u meni
od najprvog početka: sa mnom raste
u svakom času
Jednog dana
ja zastanem
a ona dalje raste
u meni dok me cijelog ne proraste
i stigne na rub mene. Moj svršetak
njen pravi je početak:
kad kraljuje dalje sama
A. B. Šimić
1. Definicija i podjela najčešćih uzroka demencije
Prema tradicionalnoj definiciji demencija (lat. de- bez, mens um) je opće, progresivno propadanje intelektualnih funkcija. Ova definicija nije dobra budući da bolesti mozga ne zahvaćaju sve njegove dijelove jednako, a biološka podloga spoznajnih procesa ima oblik preklapajućih neuronskih mreža organiziranih oko recipročno povezanih kortikalnih epicentara. Stoga se demenciju ne može promatrati kao određenu bolest, već samo kao zajednički naziv za brojne neuropsihološke promjene što nastaju kao karakteristične posljedice pojedinih kroničnih bolesti mozga (encefalopatija). Demencija se u načelu javlja kao dominantan sindrom u kliničkoj slici bolesnika kad patološkim procesom budu oštećena ključna područja sljepoočnog (entorinalni korteks, hipokampalna formacija i amigdala), čeonog (orbito- ili laterofrontalna područja), ili stražnjeg dijela tjemenog režnja. Zahvaćenost lateralne sljepoočne, prednje tjemene i inzularne moždane kore dodatno pogoršava kliničku sliku, a istodobno oštećenje projekcijskih skupina neurona crne i bazalne jezgre, lokus ceruleusa, ventralnog tegmentalnog područja i rafe jezgara dovodi i do subkortikalnih znakova i simptoma.
Velik je broj poremećaja koji mogu dovesti do oštećenja jednog ili više navedenih ključnih područja mozga. Sveukupno ima više od 50 bolesnih stanja što im demencija čini veći ili manji dio kliničke slike. S obzirom na patološke uzroke, demencija može nastati kao posljedica neurodegenerativih, vaskularnih, infektivnih (AIDS, subakutni sklerozirajući panencefalitis, herpes simplex encefalitis, neurosifilis, reaktivacija virusa ospica, itd.), traumatskih, metaboličko-toksičkih, te neoplastičkih bolesnih stanja.
S obzirom na progresiju bolesti uzroci demencije mogu se podijeliti u kronične progresivne i neprogresivne encefalopatije. Kronične neprogresivne encefalopatije nastaju obično uslijed ozljeda ili hipoksije, te rezultiraju kroničnom neprogredirajućom demencijom. Kronične progresivne encefalopatije daleko su češće, a s obzirom na uzrok dijele se u 'vanjske', metaboličke i 'unutrašnje'. Kronične progresivne encefalopatije koje nastaju uslijed 'vanjskih' uzroka (akutna difuzna ili žarišna ozljeda mozga, subduralni hematom, demencija boksača, primarni intrakranijalni tumor, metastatski karcinom, normotenzivni hidrocefalus, paraneoplastički sindrom, ciste parazita, apsces mozga, itd.) dovode do povećanog intrakranijalnog tlaka, te se relativno lako klinički razlikuju od preostalih dviju kategorija.
U metaboličke uzroke kronične progresivne encefalopatije ubrajaju se različita toksička stanja (sistemske infekcije, alkoholizam, predoziranje droga, trovanja teškim metalima i organskim otapalima), deficijencija vitamina B12, B1, B2, niacina, kronična hepatička, renalna i kardiorespiratorna encefalopatija, endokrini poremećaji (dijabetička ketoacidoza, kronična hipoglikemija, hipotiroidizam, hipo- i hiperparatiroidizam, adrenalna insuficijencija i Cushingov sindrom), bolesti nakupljanja (metakromatska leukodistrofija, Kufsova bolest, Hallervorden-Spatzova bolest), te poremećaji elektrolita (hiper- i hiponatrijemija, hiper- i hipokalcemija). Alkoholna encefalopatija u starijih muškaraca, te hipotiroidizam u starijih žena (za postavljanje dijagnoze potrebno je izmjeriti koncentraciju stimulirajućeg hormona štitne žlijezde, TSH), vjerojatno su najčešći uzroci demencije iz ove skupine. Stupanj poremetnje moždanih funkcija fluktuira u zavisnosti od vrste i relativne težine temeljne bolesti, a uobičajeni simptomi metaboličkih encefalopatija su poremećena koncentracija (smetenost), mentalna i psihička usporenost, brzo oscilirajuće stanje budnosti (npr. pospanost se izmjenjuje s povećanom nesvrsishodnom aktivnošću), anomija, nekoherentno razmišljanje (pisani jezik je uvijek jače poremećen nego govorni), poremećaji percepcije koji dovode do iluzija i halucinacija (sve do delirija u kojem ih bolesnik doživljava kao zastrašujuće), konstrukcijska apraksija, dezorijentacija (najprije u vremenu, a zatim i u prostoru, ali rijetko za vlastiti identitet), te neprikladno socijalno ponašanje. Neurološki znakovi koji često prate metaboličku encefalopatiju su tremor i mioklonus, a elektroencefalogram tipično pokazuje difuzne spore valove velikih amplituda. Za razliku od 'unutrašnjih' kronično progresivnih encefalopatija, metaboličke encefalopatije su u načelu reverzibilne, tj. mogu se izliječiti. Ovo naravno ne isključuje mogućnost da i pacijenti s 'unutrašnjom' kronično progresivnom encefalopatijom obole od metaboličke encefalopatije. Dapače, zbog smanjenih moždanih 'rezervi', u ovih su bolesnika metaboličke encefalopatije učestalije.
'Unutrašnje' kronične progresivne encefalopatije najvažnija su skupina bolesti koje uzrokuju demenciju, a s obzirom na to koji je dio mozga zahvaćen patološkim procesom dijele se na kortikalne, kortikalno-subkortikalne, subkortikalne i multifokalne. Pored Alzheimerove bolesti, najvažnije kortikalne kronično progresivne encefalopatije su žarišne (lobarne) atrofije (frontotemporalna demencija, demencija čeonog režnja, primarne progresivne afazije i agnozije), Pickova bolest, Braakova demencija i nasljedne tauopatije.
U najučestalije kortikalno-subkortikalne kronično progresivne encefalopatije ubrajaju se vaskularna demencija, bolest Lewyjevih tjelešaca i druge sinukleinopatije, alkoholna encefalopatija, kortikobazalna degeneracija.
Najvažnije subkortikalne kronično progresivne encefalopatije su progresivna supranuklearna kljenut (PSP, od 'progressive supranuclear palsy', Steele-Richardson-Olszewski sindrom), Huntingtonova bolest i Parkinsonova bolest.
U kronično progresivne encefalopatije s mnogobrojnim žarištima spadaju spongioformne encefalopatije (još se nazivaju i prionske bolesti) kao što su Creutzfeldt-Jacobova bolest (CJD, od 'Creutzfeldt-Jacob disease'), kuru, Gerstmann-Sträussler-Sheinkerov (GSS) sindrom i smrtonosna obiteljska nesanica (FFI, od ‘fatal familial insomnia’), multipla skleroza, progresivna leukoencefalopatija, neke nasljedne spinocerebelarne ataksije, amiotrofična lateralna skleroza.
Vrlo je vjerojatno da je daleko najčešći oblik demencije, bolest čije znakove i simptome suvremena medicina naziva Alzheimerovom bolešću (Alzheimer's disease, AD), postojala od davnine jer su simptome i znakove Alzheimerove bolesti opisali već stari grčki i rimski pisci, Shakespeare i drugi, iako je sam naziv za nju bio različit u različita vremena i u različitim kulturama. AD je dobila naziv po Alzheimeru, koji je bolest opisao na sastanku psihijatara jugozapadne Njemačke u Tübingenu 3. i 4. studenog 1906. godine u 51-godišnje bolesnice Auguste D.. Prvi simptomi u navedene bolesnice uključivali su promjene osobina ličnosti sa jakim osjećajem ljubomore prema mužu, a 25. studenog 1901. godine kad je dovedena u Bolnicu za duševne bolesti u Frankfurtu na Maini, imala je poremećeno pamćenje, poteškoće u čitanju i pisanju i paranoju. Kasnije su se pojavile i halucinacije, disfazija, disgnozija i dispraksija, te je naposlijetku 8. travnja 1906. godine umrla. Alzheimer je uočio i povezao navedene kliničke simptome bolesti s postmortalnim neuropatološkim obilježjima: atrofijom moždane kore i gubitkom neurona, te prisutnošću plakova i neurofibrilarnih promjena prikazanim metodom srebrne impregnacije po Bielschowskom. Njegovo je izlaganje najprije samo kratko notirano u ‘Neurologisches Zentralblatt’, a zatim i objavljeno u dva njemačka časopisa 1907. godine. 'Plakovi' su u to vrijeme već bili dobro poznati - prvi ih je opisao Beljahow 1889. godine, a zatim i Marinesco i Blocq 1892. godine, te Redlich 1898. godine u dva dementna pacijenta, što je nazvao ‘milijarnom sklerozom’ (tek je Perusini 1910. godine ovaj naziv promijenio u plakove). No, neurofibrilarnu ‘proliferaciju’ vjerojatno je prvi opisao upravo Alzheimer (iste je godine opisao i povezao s demencijom i Fischer) i to kao promjenu koju je uočio u gotovo svakom četvrtom neuronu moždane kore bolesnice Auguste D.. Kao i senilne plakove (SP) za koje je Alzheimer primijetio da se mogu prepoznati golim okom čak i bez posebnog bojanja, ovu strukturnu abnormalnost što se svjetlosno-mikroskopski očituje ponajviše neurofibrilarnim snopićima (NFT, od 'neurofibrillary tangles'), nije lako vidjeti na rutinskim hemalaun-eozinskim i krezil-violetnim (Nissl) preparatima. Prvi je vizualizaciju i SP i NFT, u mediju uklopljenim ili smrznutim rezovima, omogućio Bielshowsky razvitkom bojenja srebrnom impregnacijom 1902. godine. Ova je metoda, pored bojenja po Crossu i Gallyasu, i danas među najkorištenijima za navedenu svrhu, a upravo je pomoću nje 1911. godine sam Bielschowsky prvi uočio da se SP u središnjem dijelu sastoje od amiloida.
Nakon drugoopisanog slučaja 56-godišnjeg bolesnika s kliničkim obilježjima demencije koji je umro 1910. godine, a za kojeg je Alzheimer ustanovio prisutnost velikog broja plakova u moždanoj kori bez neurofibrilarnih promjena, Kraepelin je bio uvjeren da se radi o posebnoj bolesti, te skovao eponim Alzheimerova bolest (mozak navedenog bolesnika je nedavno ponovno analiziran modernim histološkim i molekularno-biološkim metodama: radi se o rijetkom obliku AD bez izrazitih neurofibrilarnih promjena, nije ustanovljena mutacija za amiloid prekursorni protein, a genotip apolipoproteina E bio je e3/3). Slijedeću neuropatološku značajku AD - granulovakuolarnu degeneraciju (GVD) otkrila je skupina znanstvenika koje je u Anatomskom Laboratoriju Sveučilišne Psihijatrijske Klinike u Münchenu 1903. godine okupio Alzheimer, a pripisuje se Simchowitzu. Amiloidnu angiopatiju opisao je prvi Scholz 1938. godine, a tzv. Hiranova tjelešca, Hirano 1961. godine. Nažalost, sve do današnjih dana, ni jedna se od navedenih karakterističnih neuropatoloških promjena nije pokazala specifičnom, kao niti bilo koji drugi biološki pokazatelj, jer se svi mogu naći u tijeku normalnog starenja ili različitim patološkim stanjima. Zato se može reći da ni 96 godina od prvog opisa, ne razumijemo točan uzrok i narav navedenih promjena, pa AD s pravom, pored golemog napora i velike količine prikupljenih podataka, i dalje nosi epitet ‘zagonetke svih zagonetki’. Zahvaljujući razvitku neuroanatomskih, genetskih i molekularno-biokemijskih metoda ipak se s optimizmom može reći da se sve više otkrivaju odnosi između kognitivnih promjena i njihovih morfološko-patoloških supstrata, rizični i zaštitni činitelji, te procesi koji ubrzavaju odnosnu usporavaju promjene povezane sa starenjem i nastankom bolesti.
Kroz gotovo više od pedeset godina nakon otkrića, smatrano je da se AD pojavljuje u dobi prije 65. godine života. Razlog tome bili su Kraepelinov opis AD u njegovom udžbeniku iz psihijatrije iz 1912. godine kao ‘senium precox’ ili ‘presenilna demencija’ (iako je on odmah iza toga naziva izrazio svoju sumnju pišući: ‘... moguće je da je bolest više-manje neovisna od starosti pacijenta’), te Fullerov pregledni članak o do tada opisanim slučajevima AD i jednom vlastitom, gdje je svih trinaest bolesnika umrlo u dobi mlađoj od 65 godina. Dok se 'presenilna demencija' izjednačavala s AD, izraz 'senilna demencija' korišten je za opis prisutnosti demencije u starijih osoba i također se primjenjivao bez obzira na uzrok, npr. i za AD i za cerebrovaskularnu bolest. Radi isticanja razlike u odnosu na ‘presenilnu demenciju’, skovan je potom izraz 'senilna demencija Alzheimerovog tipa'. Danas se više ne bi trebao koristiti ni jedan od navedenih izraza jer AD može nastupiti u bilo kojoj dobi poslije 30. godine života (najmlađi dokumentirani bolesnik imao je 26. godina), a osim AD i cerebrovaskularnih bolesti postoje i mnogi drugi uzroci kako ‘presenilne’ tako i ‘senilne demencije’.
Podmukla i postepena progresija oštećenja spoznajnih funkcija glavno je kliničko obilježje AD, a slabljenje zapamćivanja najizraženiji i najstalniji znak. Simptomatologiju AD prvi je 1950. godine detaljno opisao Sjögren, te je podijelio u 3 stadija. U prvom, ranom stadiju, karakterističan je gubitak epizodičkog deklarativnog pamćenja, naročito usvajanja novih sadržaja, što je praćeno i progresivnom propadanjem mogućnosti prisjećanja već usvojenih epizodičkih sadržaja (ovakav obrazac oštećenja pamćenja sličan je onome kakav se vidi nakon obostranog odstranjenja hipokampusa). Poteškoće u pronalaženju prave riječi (anomička disfazija) u tijeku spontanog govora obično su prvi karakteristični znak uslijed čega pacijent zaobilaznim putem nastoji reći što je naumio (logoreja). Oscilacije raspoloženja s promjenenama u osobinama ličnosti mogu biti dodatni simptomi. Bolesnik može imati probleme u izvođenju svakodnevnih aktivnosti, npr. oblačenju, vođenju evidencije čekova, pronalaženju željenog puta, ili prisjećanju gdje mu se nalaze stvari. Može biti oštećeno ispravno procjenjivanje i apstraktno mišljenje, prepoznavanje osoba (prosopagnozija), a u većine se uočava nedostatak spontanosti i inicijative. Kao prvi symptom, u oko 10% bolesnika mogu se javiti i izolirana afazija ili vremenska i prostorna dezorijentacija. Također se mogu vidjeti agresivnost i iritabilnost, obično kao posljedica gubitka kontrole i sposobnosti zapamćivanja. Ovi se simptomi ipak javljaju u manjeg broja bolesnika, jer većina uglavnom nije svjesna propadanja vlastitih spoznajnih sposobnosti. U manjem broju slučajeva već u ovom ranom stadiju mogu biti prisutne halucinacije i paranoidne ideje, a neurološki status je obično uredan. Rezultat MMSE (od ‘Mini-Mental State Examination’) obično je između 18 i 26 (od mogućih 30 bodova; odgovor na svako pitanje donosi 1 bod).
U drugom stadiju jasno je izražena progresija demencije, te sve više dolaze do izražaja i žarišni neokortikalni simptomi: apraksija, agnozija i afazija. Govor i čitanje sve su lošiji, dok je sama artikulacija obično još sačuvana. Učenje, prepoznavanje najbliže okoline i rodbine, te semantičko pamćenje postaju sve slabiji, dok pamćenje davnih događaja ne mora biti u potpunosti izgubljeno. Rezultat MMSE je između 10 i 18. Još uvijek je dobro sačuvano proceduralno pamćenje, tj. sposobnost učenja novih motoričkih obrazaca. Zbog gubitka osjećaja o prostoru i vremenu, agitacija, agresivnost i nekooperativnost sve više napreduju, uslijed čega se mogu razviti i depresija, anksioznost i paranoidne reakcije. Deluzije, halucinacije i neprimjereno seksualno ponašanje javljaju se manjem broju slučajeva. Česta je i pospanost po danu, te zbunjenost nastupanjem sumraka (‘sundowning’). Mogu nastupiti i znakovi rigidnosti (ali bez hiperkinezije), generalizirani epileptički napadaji i logoklonus.
U posljednjem, trećem stadiju, klinička je slika više jednolična, ali istovremeno i difuznija s obzirom na žarišne znakove. Rezultat MMSE je manji od 10. Deklarativno pamćenje je u potpunosti izgubljeno (i epizodičko i semantičko), a počinje se gubiti i proceduralno. Svakodnevne aktivnosti postaju nemoguće jer bolesnik više ne može hodati, žvakati, gutati, kontrolirati sfinktere, te postaje potpuno ovisan o skrbi drugih. Ovaj stupanj ovisnosti testira se i mjeri upitnikom o svakodnevnim aktivnostima (ADL, od 'Activities of Daily Living questionnaire'). Javljaju se motorički i drugi žarišni neurološki ispadi, a također raste i incidencija epileptičkih napadaja. Prevladavaju znakovi oštećenja čeonog režnja: primitivni refleksi, paranoja, disinhibicija, fleksijske kontrakture, stereotipni pokreti i ponavljanje istih fraza, riječi ili slogova. Naposlijetku nastupaju koma i smrt, obično od infekcije. Sjögrenov klinički opis vrijedi i danas, iako su od tada brojni autori doprinijeli još detaljnijem i točnijem opisu složene i vrlo varijabilne kliničke slike.
Tako je opisano da se depresivni i psihotični simptomi javljaju i u do 30-40% bolesnika, od čega su paranoidne misli najčešći psihotički simptom, a javljaju se prosječno u oko 16% slučajeva. U jednoj većoj studiji agresivno je ponašanje zabilježeno u 52% bolesnika, od toga ih je 35% bilo samo verbalno agresivno, a 18% i fizički. Vidne halucinacije susreću se u oko 13%, a slušne u oko 10% bolesnika. Halucinacije su obično povezane s bržim kognitivnim propadanjem. Čini se da bolesnici s depresivnim simptomima i schizophrenijom u prosjeku imaju slabija kognitivna oštećenja i manje proširen ventrikulski sustav, što je možda povezano s uzimanjem antidepresivnih i antipsihotičkih lijekova. Bolje raspoloženje od onoga prije nastanka bolesti zabilježeno je u 3.5% do 17% pacijenata. Ekstrapiramidni sindrom javlja se u 9% do 36%, mioklonus u 5% do 55%, a generalizirani kloničko-tonički napadaji u 9-64% bolesnika. Smatraju se kasnim znakom AD, a povezani su s velikim gubitkom neurona zahvaćenih dijelova korteksa.
Biološki pokazatelji AD mogu se podijeliti na središnje (intrakranijalne) i periferne. Do 1980. godine veći je dio istraživača mislio da je AD ograničena na mozak, dok su samo neki pretpostavljali da se radi o sistemskoj bolesti. Tada su u bolesnika s AD otkrivene tri različite abnormalnosti u perifernim tkivima: povećana aktivnost monoaminooksidaze u trombocitima, nestabilnost kromosoma u limfocitima i abnormalnosti membrane eritrocita. Kasnije su opisani i pojačani odgovor na oralni test tolerancije glukoze s gubitkom tjelesne težine usprkos odgovarajućoj prehrani, abnormalnosti fibroblasta, smanjeni broj CD8+ limfocita T, smanjen broj muskarinskih i nikotinskih receptora na limfocitima i drugi poremećaji. Ipak, od svih pronađenih sistemskih učinaka AD niti jedan se nije pokazao specifičnim, a ovi nalazi su još više naglasili pitanje zašto bolest selektivno zahvaća baš mozak? Odgovor je očigledno je u direktnom odnosu sa etiologijom bolesti, koja je još uvijek nedovoljno poznata.
Mogući neurokemijski kandidati za biološke pokazatelje AD uključuju neurotransmitere, neuropeptide i aminokiseline. Brojna su istraživanja ustanovila deficit aferentnih kortikalnih sustava koji kao načelni neurotransmiter koriste acetilkolin, dopamin, noradrenalin i serotonin. Jedan od najstalnijih nalaza je zahvaćenost kolinergičkog sustava, iz čega se početkom osamdesetih godina razvila tzv. 'kolinergička hipoteza poremećaja pamćenja u starenju i AD'. Naime, najprije je otkrivena redukcija acetilkolin-esteraze (AChE), a zatim i smanjenje aktivnosti kolin-acetiltransferaze (ChAT) u neokorteksu i hipokampusu bolesnika s uznapredovalom AD. Na to su se nadovezali dokazi o degeneraciji glavnog izvora kolinergičke inervacije korteksa, bazalne Meynertove jezgre, te degeneraciji glavnog izvora kolinergičke inervacije hipokampusa, medijalne septalne jezgre. U cerebrospinalnoj tekućini oboljelih od AD nađena su protutijela na kolinergičke neurone, smanjena aktivnost AChE i smanjena koncentracija kolina. No, zbog preklapanja vrijednosti s nedementnim osobama kolinergički deficit se ne može pouzdano utvrditi. Čini se da smanjeni broj nikotinskih receptora u moždanom tkivu relativno dobro korelira sa stupnjem demencije.
Od neuropeptidergičkih pokazatelja, u moždanom je tkivu najčešći nalaz smanjena razina somatostatina, iako ni ovaj pokazatelj nema dijagnostičku vrijednost, jer se redukcija somatostatina viđa u brojnim psihijatrijskim i neurološkim poremećajima. Istraživanja koncentracija aminokiselina u cerebrospinalnoj tekućini bolesnika s AD većinom nisu podudarna, ali u skladu s novijim shvaćanjima (vide infra) ističe se važnost nalaza povećane razine glutamata i prolina, te smanjene razine taurina, ornitina i lizina.
Razvitak tehnika za vizualizaciju moždanog tkiva i aktivnosti moždanih stanica (MRI, fMRI, PET, SPECT, rCBF) otvorio je nove načine za dijagnostiku i istraživanje AD, iako je još uvijek glavna ‘svrha’ ovih metoda otkrivanje reverzibilnih ('vanjskih') uzroka demencije. Atrofija hipokampusa uobičajeno je obilježje uznapredovalih slučajeva AD i relativno dobar radiološki pokazatelj prisutnosti bolesti. Npr. u jednoj velikoj studiji nađena je u 87% bolesnika s izraženom AD. No, blaža redukcija volumena hipokampusa postoji i u klinički normalnih starijih osoba. Dobru pozitivnu korelaciju između patoloških promjena u AD pokazuje također dilatacija hipokampalne fisure. Više skupina istraživača pokušalo je na temelju nalaza WMH (WMH, od 'white matter hyperintensities') razviti dijagnostički kriterij pomoću kojeg bi se magnetskom rezonancijom razlikovale dementne od nedementnih osoba. No, WMH su bili prisutni u samo 30-60% bolesnika s AD, a nađeni su i u normalnih nedementnih osoba. Nadu u dijagnostičku vrijednost WMH ipak daje nalaz da je oblik periventrikularnih WMH (a ne onih u dubokim dijelovima bijele tvari) puno češći u bolesnika s AD, nego u ostalih skupina ispitanika, npr. onih s vaskularnom demencijom (VaD), jer se u AD WMH javljaju ponajviše u periventrikularnom području čeonih rogova lateralnih ventrikula. Stupanj WMH pozitivno korelira sa stupnjem kognitivnog oštećenja u AD. Istraživanja cerebralnog metabolizma pomoću pozitronske emisijske tomografije pokazala su da je obostrani hipometabolizam sljepoočnih i tjemenih dijelova mozga najranija karakteristična abnormalnost u AD (osjetljivost oko 92%, specifičnost 80%). U umjereno do jako dementnih bolesnika uočava se i zahvaćenost čeonih režnjeva. SPECT-om se obično ustanovi smanjen protok krvi u sljepoočnim područjima mozga.
Prvi pokušaj u olakšavanju dijagnosticiranja AD bilo je uvođenje ljestvice od 12 kliničkih obilježja 1982. godine. Svakom obilježju pridani su bodovi, i to po 2 boda za: amneziju, prostornu dezorijentaciju, apraksiju/agnoziju/afaziju, logoklonus i povećani mišićni tonus, te po 1 bod za: sporu progresiju simptoma, gubitak pronicljivosti i opažanja, logoreju, progresivnu redukciju spontanog govora, epileptički napadaj, miokloničke grčeve i Klüver-Bucy sindrom. Ova ljestvica ne uključuje neurofiziološke, psihometrijske i podatke dobivene snimanjem mozga. Pa ipak, budući da je podudarnost simptomatologije različitih slučajeva AD relativno visoka, postoji pozitivna korelacija zbroja dobivenog prema ljestvici i postmortalne neuropatološke dijagnoze AD. Glavna poteškoća je u tome što ima i velik broj lažno negativnih rezultata, što znači da se prema ovoj ljestvici dijagnoza AD donosi puno rjeđe nego što bi to trebalo s obzirom na prevalenciju AD u neuropatološkim izvještajima.
Kako nije pronađen ni jedan zadovoljavajući biološki pokazatelj za postavljanje dijagnoze AD, i danas se klinička dijagnoza temelji na simptomatologiji. Tri međunarodno prihvaćena kriterija su: DSM-IV (od ‘Diagnostic and Statistical Manual of Mental Disorders’, četvrto izdanje), NINCDS-ADRDA kriterij (od ‘National Institute of Neurological and Communicative Disorders and Stroke and Alzheimer's Disease and Related Disorders Association’) i ICD-10 (od ‘International Classification of Disease’, deseta revizija).
U DSM-IV kriteriju, pored kognitivnih deficita kao što su oštećenje pamćenja, afazija, agnozija, apraksija, navodi se da deficit mora biti dovoljan da dovede do poremećaja normalnog djelovanja osobe na radnom mjestu i u društvu. Pouzdanost (specifičnost) dijagnoze prema ovom konceptu iznosi 0.55-0.65, a osjetljivost od 0.71-0.8.
Koncept NINCDS-ADRDA Radne Skupine, temelji se na slijedećim glavnim kriterijima: 1. demenciji ustanovljenoj kliničkim ispitivanjem, dokumentiranoj MMSE ili nekim sličnim testom, te potvrđenoj neuropsihološkim testiranjem, 2. deficitima u dva ili više kognitivnih područja, 3. progresivnom slabljenju pamćenja, i 4. odsutnosti sistemskih poremećaja ili drugih bolesti koje zahvaćaju mozak. Ovaj koncept uključuje i kategorizaciju vjerojatnosti točnosti postavljanja dijagnoze u 3 stupnja (skoro sigurna, vjerojatna i moguća dijagnoza AD). Pouzdanost dijagnoze prema ovom konceptu između različitih institucija varira od 0.49-0.64.
Prema ICD-10, AD uključuje prisutnost demencije, podmukao početak, sporu deterioraciju i odsustvo drugih bolesti koje mogu uzrokovati demenciju. Dakle, sva tri navedena kriterija imaju slijedeće zajedničke uvjete: bolesnik mora biti dementan, imati oštećenje pamćenja, te oštećenja u drugim kognitivnim domenama, a moraju biti odsutne druge bolesti koje uzrokuju demenciju. NINCDS-ADRDA i ICD-10 isključuju dijagnozu AD u prisutnosti žarišnih neuroloških znakova, dok DSM-IV isključuje dijagnozu AD ako su prisutne depresija i schizophrenia.
Često se ponavlja pitanje koji od navedenih kriterija daje najbolje rezultate u odnosu na postmortalni neuropatološki nalaz. Opće je prihvaćena činjenica da DSM-IV ima bolju specifičnost, dok NINCDS-ADRDA pokazuje veću osjetljivost (86-100% slučajeva koji klinički zadovoljavaju navedeni kriterij bude potvrđeno neuropatološki). Čini se da je najveće ograničenje NINCDS-ADRDA kriterija nedovoljno jasna stavka o prisutnosti žarišnih neroloških znakova ili simptomatologije Parkinsonove bolesti. Poboljšanja kliničkih kriterija u budućnosti bit će usmjerena prema preciznijem kliničkom opisu simptoma bolesti i još detaljnijem neuropsihološkom testiranju podešenom prema spolu, starosti i stupnju edukacije.
Heterogenost i varijabilnost kliničkih obilježja, kao i histopatološkog nalaza, navela je mnoge autore na pomisao o klinički različitim podvrstama AD. Najčešća je podjela na bolesnike s ranim i bolesnike s kasnim početkom AD (poslije 65. godine života). Mjereno duljinom trajanja bolesti, AD obično ima brži tijek u mlađih bolesnika, a afazija, apraksija i agnozija u srednjem stadiju bolesti također su karakterističnije za slučajeve s ranijim početkom bolesti. Zbog ogromne individualne varijabilnosti i šarolikosti kliničke slike, ni jedna od kliničkih podjela AD nije se pokazala korisnom niti upotrebljivom u prognozi bolesti. S dijagnostičkog stajališta, problem je u tome da se navedeni simptomi ne javljaju u isto vrijeme, niti u istom slijedu. Nadalje, oni variraju u jačini ili pak mogu biti grupirani na različite načine, što također doprinosi heterogenosti kliničke slike. Kad se bolesnici s AD promatraju kroz dulje razdoblje, redovito se susreću i asimetrije u zahvaćenosti lijeve i desne hemisfere. Navedene razlike također ne moraju značiti da se radi o različitim podvrstama AD, već bi ih prije trebalo pripisati nejednakomjernoj progresiji bolesti.
Pored MMSE, danas se koriste i brojni drugi (bolji) psihometrijski testovi za rano otkrivanje, klasificiranje i praćenje kognitivnih promjena u AD, npr. ADAS-cog (‘Alzheimer's Disease Assessment Scale – cognition’), ‘Blessed Information-Memory-Concentration’ (BIMC) - dio je BDS (od ‘Blessed Dementia Scale’) testa za utvrđivanje stupnja demencije i ‘Clinical Dementia Rating’ test (CDR). Izvrsnu linearnost sa stupnjem kognitivne deterioracije daju Bentonov test vidne retencije (BVRT), ‘Boston Naming Verbal Fluency’ test (BNVFT), BIMC i ADL. Jedan od potencijalno najvažnijih psihometrijskih testova za rano ustanovljavanje AD je BVRT, jer s visokom osjetljivošću i specifičnošću predviđa AD i prije nastanka očiglednih kognitivnih simtoma. BVRT zahtijeva od ispitanika da reproducira geometrijske oblike nakon što ih je gledao kroz 10 sekundi. Dakle, testira se kratkotrajno pamćenje, a rezultat na testu se predstavlja kao ukupni broj grešaka.
Dijagnosticiranje AD testiranjem povećane osjetljivosti pupile na djelovanje kolinergičkih antagonista nije se pokazalo dovoljno dobrom metodom, jer predstavlja nespecifični znak kolinergičke preosjetljivosti.
U diferencijalnoj dijagnozi AD dolaze najprije u obzir sve druge kortikalne kronično progresivne encefalopatije, a tek onda i ostali uzroci demencije. Od ‘unutrašnjih’ kronično progresivnih encefalopatija najveće dijagnostičke poteškoće zadaju vaskularna demencija i bolest Lewyjevih tjelešaca, a glavni razlog je činjenica da ove dvije bolesti mogu postojati istovremeno s AD (npr. u jednom većem istraživanju vaskularna demencija je bila prisutna u 45% bolesnika s AD). Pored navedenih bolesti u užu diferencijalnu dijagnozu AD najčešće dolaze i alkoholna encefalopatija, hipotireoza, dobroćudna staračka zaboravljivost (BSF, od ‘benign senile forgetfulness’, naziva se i ‘age-associated memory impairment’: predstavlja slabo progresivno ili neprogresivno slabljenje kratkotrajnog pamćenja koje prati ‘normalno’ starenje), te sekundarne demencije uslijed drugih neuroloških (karcinomatozni meningitis, paraneoplastikni encefalitis, amiotrofička lateralna skleroza, multipla skleroza, nasljedne ataksije, itd.) i psihijatrijskih poremećaja (shizophrenia). Također treba imati na umu i depresiju, budući da depresivni bolesnici često pokazuju znakove demencije (pseudodemencija), što se katkad teško mogu razlikovati od prave demencije. Ipak, za razliku od AD, gdje bolesnik nastoji minimalizirati svoje deficite, depresivni bolesnici ih obično uvećavaju, a npr. afazički deficiti kao što su parafazije gotovo se nikad ne vide u depresivnih pacijenata. Samo mali broj depresivnih bolesnika razvije sliku pseudodemencije, dok relativno veliki broj dementnih bolesnika također bude i depresivan (odnosno abuličan ako je patološkim procesom zahvaćen čeoni režanj).
Neurodegenerativne bolesti koje dovode do demencije po zdravstvenim (morbiditet i mortalitet su im odmah iza kardiovaskularnih i malignih tumorskih bolesti), socijalnim (onemogućavaju kvalitetan život oboljele osobe u zajednici) i ekonomskim pokazateljima (troškovi uzdržavanja i liječenja oboljelih od AD u zapadnome će svijetu uskoro biti jednaki kao zajednički troškovi za kardiovaskularne bolesti, rak i moždani udar zajedno) imaju danas ogroman značaj. Zbog porasta udjela starije populacije u budućnosti se očekuje daljnje povećanje broja oboljelih (u Švedskoj se npr. očekuje porast od 50% u razdoblju od 1991-2025. godine: s 154 na 234 tisuće oboljelih). AD je sveukupno najčešći (50-70%) oblik demencije u svim zemljama svijeta u kojima je ova bolest detaljno proučavana (a nakon 65 godina starosti AD je uzrokom demencije u više od 80% dementnih bolesnika). Prevalencija varira, ali u prosjeku se može reći da je po učestalosti AD četvrti uzrok smrti u zemljama zapadnog svijeta. Od AD obolijeva 3-10% populacije ispod 65 godina starosti, te 25-50% populacije iznad 85 godina starosti.
Epidemiološki su utvrđeni mnogobrojni rizični činitelji za nastanak bolesti, iako način njihovog utjecaja nije poznat. Rizični činitelji se dijele na definitivno prihvaćene (relativni rizik veći od 2, što znači da je toliko puta veća vjerojatnost za nastanak bolesti), upitne (relativni rizik između 1.5 i 2), moguće rizične činitelje (relativni rizik veći od 1), te zaštitne činitelje (relativni rizik manji od 1). U definitivno prihvaćene spadaju rizične činitelje spadaju: 1. Dob. Ovo je najznačajniji rizični činitelj. Prevalencija AD udvostručava se svakih 5.1 godina u osoba starosti između 65 i 85 godina, dakle gotovo eksponencijalno; 2. Oba e4 alela za apolipoprotein E (relativni rizik oko 2.27); 3. Pozitivna obiteljska anamneza (relativni rizik oko 3.2). Ovo ne znači da je a priori razlog genetski, već on može biti i okolišni (npr. nekad se mislilo da je tuberkuloza nasljedna bolest). Nažalost ni jedan od navedenih rizičnih činitelja ne može se (zasad) iskoristiti za prevenciju nastanka AD. U još uvijek upitne rizične činitelje spadaju: 1. Spol. Žene sveukupno češće obolijevaju od AD (iako prije 70 godina starosti češće obolijevaju muškarci). Ta proporcija se naročito povećava poslije 75. godine života, da bi do 90. godine prema nekim istraživanjima dostigla i 9:1 (upravo obrnuto od VaD), ali je za to vjerojatno djelomično odgovorna i činjenica da žene u prosjeku žive duže nego muškarci. Ovaj podatak također ukazuje da ženski spolni hormoni možda imaju zaštitnu ulogu u nastanku AD (relativni rizik za estrogen je oko 0.4); 2. Stupanj školske spreme. Osobe nižeg stupnja edukacije ili zaposlenja imaju gotovo dva puta veću vjerojatnost obolijevanja od AD, te skoro tri puta ako su istovremeno i nižeg stupnja edukacije i nižeg stupnja zaposlenja. Pretpostavlja se da niži stupanj edukacije i zaposlenja onemogućuje ovim bolesnicima bolje podnošenje početnih oštećenja u tijeku bolesti, budući da imaju manju ‘sinaptičku rezervu’. Alternativno je moguća i povezanost navedenih varijabli s nižim socioekonomskim i nutritivnim statusom; 3. Trauma glave. Jedna od brojnih pretpostavki je oštećenje mikrocirkulacije uslijed udarca ili čestih trauma glave, s hematogenim ulaskom beta-amiloida u mozak. Pored navedenih predloženi su (ali nepotvrđeni) i brojni drugi, manje značajni rizični činitelji, kao što su mutacije i polimorfizmi a2-makroglobulina i LRP (od ‘lipoprotein-related protein’) receptora (što ujedno djeluje i kao receptor za odstranjivanje APP i ApoE), angiotenzin-1 konvertirajućeg enzima, a1-antikimotripsina, bleomicin hidroksilaze, katepsina D, dihidrolipoil sukciniltransferaze (DLST), gena estrogenskog receptora a, gena HLA sustava, lipaze lipoproteina, mijeloperoksidaze, neurotrofina-3, endotelijalnog oblika sintaze dušičnog monoksida, transportera ponovnog unosa serotonina, VLDL receptora, te smanjena koncentracija testosterona, uzimanje prekomjernih količina alkohola, povećani krvni tlak i povećana koncentracija homocisteina u plazmi, a od 'zaštitnih' činitelja ističu se pušenje i uzimanje nesteroidnih protuupalnih lijekova (relativni rizik oko 0.4), ali ne acetilsalicilne kiseline.
Obiteljsko pojavljivanje (FAD, od 'familial AD') činitelj je rizika za nastanak AD. Prvi dobro dokumentirani primjer FAD opisao je Schottky 1932. godine. Od tada je do danas opisano više od stotinu vrsta FAD. Neki istraživači vjeruju da je genetska komponenta neophodna čak i u sporadičnih slučajeva AD. Prema do danas poznatim epidemiološkim podacima, obiteljski se AD javlja u oko 10-45% slučajeva, dok je ostala većina slučajeva sporadična, odnosno u njih (još) nije otkrivena genetska podloga nastanka AD.
Najprije je 1991. godine otkrivena povezanost FAD s mutacijom gena za APP (amiloid prekursorni protein) na 21 kromosomu (tzv. AD tip 1), a zatim 1992. godine i s mutacijom gena za presenilin 1 (PS-1) na 14 kromosomu (AD tip 3). Oba su ova oblika povezana s ranim početkom bolesti (između 40 i 60. godine starosti). Zatim je 1993. godine otkriven lokus ApoE gena na 19-tom kromosomu povezan s kasnim početkom AD (AD tip 2), 1995. godine 1-om (presenilin-2, PS-2) kromosomu (AD tip 4) povezan s ranim nastankom bolesti u njemačkih obitelji u dolini rijeke Volge, te 1997. godine na 3-ćem kromosomu (gen za K varijantu enzima butirilkolinesteraze, K-BChE) povezanim s kasnim početkom bolesti. Također 1997. godine otkriven je i nasljedni oblik AD zbog heretoplazmičkog (maternalnog) nasljeđivanja mutacija citokrom oksidaze I i II (AD tip 5). Iz ovih se podataka vidi da je AD i po genetskoj slici vrlo heterogena bolest. Novija genetska istraživanja ukazuju da postoji još najmanje 5 gena (na 9, 10, 11 i 12 kromosomu) povezanih s AD, od kojih jedan ima vjerojatno i jači utjecaj od apoE na dob pojavljivanja bolesti (gen za enzim koji razgrađuje inzulin na 10 kromosomu).
Trajanje i dob početka bolesti varira, a to se odnosi i na obitelji s poznatom mutacijom gena. Jedna od većih studija FAD (180 dementnih osoba iz 24 obitelji) navodi prosječno trajanje bolesti od 9 godina, s rasponom od 1 do 23 godine.
Gen za apolipoprotein E (apoE) pokazuje izraženi polimorfizam jer ima tri različita oblika: e2 (112. i 158. aminokiselina je cistein), e3 (na poziciji 112 je cistein, a na 158 arginin) i e4 (na obje pozicije je arginin). Najčešći je oblik e3. ApoE4 se javlja u otprilike 40%-65% svih FAD s kasnim početkom bolesti, a ovaj nalaz nije ograničen na FAD, nego pokazuje pozitivnu povezanost i sa sporadičnim slučajevima AD (pogotovo onima koji su imali traumu glave). Izračunato je da osobe koje naslijede oba e4 alela, imaju najmanje osam puta veću vjerojatnost za nastanak AD u odnosu na osobe s e3/3 genotipom. Npr. osoba koja ima 78. godina i obje kopije apoE4 gena ima 98%-tnu vjerojatnost obolijevanja od AD, s jednom kopijom 60%-tnu, a bez i jedne kopije samo 25%-tnu vjerojatnost. Bolesnici s oba apoE4 gena u prosjeku počnu pokazivati simptome AD prije 70-te godine života, dok oni koji nemaju apoE4 gena u prosjeku obolijevaju poslije 80. godine života. Iz ovih podataka proizlazi da apoE4 na neki način ubrzava proces nastanka AD. Čini se da najrjeđi oblik, apoE2, još više smanjuje rizik obolijevanja, tako da osobe s jednim E2 i jednim E3 genom imaju samo četvrtinu vjerojatnosti obolijevanja u odnosu na osobe s oba E3 gena. Nažalost, iako je apoE4 zasad najbolji biološki marker za prognozu napredovanja bolesti, na temelju njegove prisutnosti u uzorku krvi nije moguće predvidjeti nastanak AD, jer postoje i zdrave osobe koje imaju apoE4, te osobe koje nemaju apoE4 alel, a ipak obole od AD (ni osjetljivost ni specifičnost ne prelaze 50%). Stoga pretraživanje (‘screening’) na apoE4 ne bi doveo do željenih rezultata, naročito zbog činjenice što bi bilo previše lažno pozitivnih rezultata. Ipak, apoE genotipizacija, pored ostalih kliničkih nalaza, pomaže donekle u donošenju kliničke dijagnoze bolesti.
Nakon prepoznavanja uloge genetičkih činitelja u etiopatogenezi AD postavilo se pitanje sličnosti kliničkih obilježja u sporadičnih i obiteljskih slučajeva. Općenito se smatra da se FAD od sporadičnih slučajeva ne mogu razlikovati ni po kliničkoj niti po neuropatološkoj slici, čak ni u slučajevima s autosomno dominantnim nasljeđivanjem. Katkad se ipak u nekih obitelji motorički simptomi javljaju ranije no što je uobičajeno, a u nekih se javljaju epileptički napadaji kojima prethode mioklonički tikovi kroz nekoliko godina. Zbog velike sličnosti između sporadičnih i obiteljskih slučajeva, obiteljski su slučajevi izvrstan model za longitudinalne studije, a relativno slična dob obolijevanja u FAD unutar obitelji također omogućuje promatranje stadija AD i prije izraženih simptoma bolesti. Pa ipak, konkordancija AD je i u monozigotnih (59-67%) i dizigotnih (22-40%) blizanaca relativno niska. Čak i kad oba blizanca obole, ponekad i vrlo dugačak razmak između obolijevanja, te velika različitost patoloških promjena unutar porodica s dominantnim naslijeđivanjem AD, upućuju na značajnu uključenost činitelja okoline u patogenezi bolesti.
Najizraženije makroskopske promjene mozga u AD uključuju smanjeni volumen i težinu mozga (obično ispod 1100 grama), simetrično i difuzno atrofične vijuge s posljedično proširenim brazdama - najviše u sljepoočnim i čeonim režnjevima (u nekih bolesnika mogu biti jače zahvaćeni i tjemeni i eventualno zatiljni režnjevi, ali su subkortikalne strukture, bazalni gangliji, mali mozak i moždano deblo gotovo uvijek sačuvani), proširene lateralne moždane komore - ponajviše sljepoočni rog (hydrocephalus ex vacuo), skvrčenu bijelu tvar, smanjen i izblijedio locus coeruleus (ponekad i do mjere da se ne može ni naći), atrofirani bulbus i traktus olfaktorijus, zadebljana dura mater (histološki odgovara povećanom broju fibroblasta i kolagena) - naročito u području medijane linije. U mlađih bolesnika atrofija moždane kore odnosi se prvenstveno na smanjenje debljine korteksa, dok je u starijih očitije smanjenje kortikalne površine (a ne toliko debljine korteksa), što možda odražava kolumnarni gubitak neurona i neuropila. Općenito govoreći, atrofija moždane kore, proširenje ventrikula i druge patološke promjene izraženije su u mlađih bolesnika.
2.6.1. Senilni plakovi i amiloidna kaskadna hipoteza
Iako se već desetljećima nazivaju senilnim plakovima (SP, od ‘senile plaques’), ove se promjene nalaze i u preseniju, kako u bolesnika s AD, tako i u normalnih osoba. Započinju kao difuzni ('preamiloidni') SP (DP, od 'diffuse plaques') u kojima beta-amiloid (Ab, od ‘amyloid beta’) nema pravilnu konformaciju. Oko DP nema reakcije glija stanica, a gotovo su neizbježna posljedica starenja. Rutinski se prikazuju bojanjem metenaminom, Campbell-Switzerovom metodom, protutijelima na Ab i drugim metodama. Depoziti amiloida mogu se naći i oko krvnih žila, ispod pijalne površine ili radijalno orijentirani prema plakovima. S vremenom DP prelaze u ‘primitivne’ SP, koji sadrže guste snopiće Ab s konformacijom beta uzvojnice. Zbog biofizičkih osobina beta uzvojnice za vizualizaciju NP (ali ne i DP) koriste se Kongo crvenilo i tioflavin S bojenje. S pojavom distrofičnih neurita (DN, od ‘dystrophic neurites’) u području plaka nastaju neuritički SP (NP), koji predstavljaju glavno neuropatološko obilježje AD. Stvaranjem gustog središta od Ab nastaju ‘zreli’ SP. Iako su NP vrlo heterogeni, obično se dijele na distrofične NP s fuziformnim DN, degeneriranim dijelovima sinaptičkih elemenata i lizosomalnim gustim slojevitim tjelešcima, a bez sparenih uzvojitih filamenata (PHF, od ‘paired helical filaments’), te NP koji sadrže PHF. Distrofični tip NP se nalazi i u AD i normalnih starijih ljudi, kao i drugih sisavaca, može se ‘proizvesti’ u transgeničnih miševa i ne korelira s kognitivnim propadanjem, dok se PHF-tip nalazi isključivo u ljudi, naročito u AD (u manjoj mjeri još se može naći samo u Guam demenciji s parkinsonizmom, progresivnoj supranuklearnoj kljenuti, kortikobazalnoj degeneraciji i frontotemporalnoj demenciji). Distrofični tip može, ali i ne mora prethoditi PHF-tipu NP. Neuriti PHF-tipa u odsutnosti SP nazivaju se neuropilnim nitima (NT, od ‘neuropil threads’). Vjerojatno uslijed defosforilacije i razgradnje PHF, mogu nestati neuritički elementi plaka, tako da od NP ostanu samo gusto zbijeni amiloid, te glijalni i neuronalni nastavci – to je tzv. 'izgorjeli’ plak (‘burnt-out plaque’). Pored fibrilarnog Ab i DN, NP su (za razliku od DP) okruženi i proliferirajućim, reaktivnim astrocitima i mikroglijom, a kako se DP nalaze na mjestima gdje se nikada ne nađu zreli NP (npr. u kori malog mozga i bazalnim ganglijima), pretpostavlja se da na nastanak utječu još neotkriveni, mikrookolišni činitelji. Neki istraživači vjeruju da razlika u tipu plakova postoji od samog početka, odnosno da proces nastanka plaka započinje od degeneriranih vlakana.
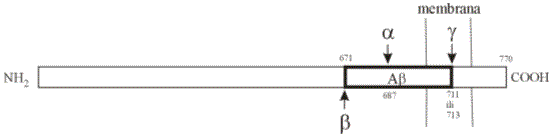
Beta-amiloid sastoji se od 39-43 aminokiseline, a molekularna težina mu je 4.2 kDa. Nastaje iz prekursorne molekule što je po njemu nazvana prekursorni protein za beta-amiloid (AbPP, od 'amyloid-beta precursor protein'). Prije odcjepljivanja, u molekuli AbPP amiloidni protein zauzima tzv. beta-A4 područje, koje se nalazi djelomično u izvanstaničnoj, a djelomično u transmembranskoj domeni AbPP (Shema 1). Zato se amiloidni protein naziva još i beta-A4 amiloid ili skraćeno samo beta-amiloid (Ab). Normalno sintetizirani APP je 110-135 kDa teška integralna transmembranska molekula glikoproteinske strukture, s velikim izvanstaničnim dijelom i malim citoplazmatskim repom (Shema 1). APP je član velike porodice visoko konzerviranih proteina, u koju još spadaju APLP1 i APLP2 (od ‘amyloid precursor protein-like protein’), no oni ne kodiraju Ab. Deset različitih oblika APP nastaju naizmjeničnim sparivanjem 19 eksona APP gena (najkraći ima 563, a najdulji 771 aminokiselinu). Samo tri transkripta sadrže Ab slijed, a kodiraju APP duljine 695, 751 i 770 aminokiselina (AbPP695, AbPP751 i AbPP770). Jedino se AbPP695 eksprimira isključivo u mozgu. Prvi je Ab u krvnim žilama primijetio Scholz, a upravo su iz vaskularnog amiloida Glenner i Wong 1984. godine otkrili njegovu primarnu aminokiselinsku strukturu.
Shema 1. Shematski prikaz AbPP770 (lokus gena 21q21.3-q22.05). Za objašnjenje vidi tekst.
Glasnička RNA za APP pronađena je u gotovo svim tjelesnim stanicama, osim granulocitima. APP putuje i anterogradnim i retrogradnim aksonskim transportom, a na presinaptičkom dijelu sinapse dolazi u interakciju s molekulama izvanstaničnog matriksa. Njegova sličnost s proteazom neksinom II ukazuje na ulogu u regulaciji i rastu aksona. Budući da relativna količina APP u stanicama može biti promjenjena aktivacijom različitih receptora i signalnih mehanizama koji uključuju protein kinazu C, ione kalcija i metabolite arahidonske kiseline, te da se aktivno procesira za vrijeme neurotransmisije, čini se da ima važnu ulogu u sinaptičkoj plastičnosti. Visoka konzerviranost i citoplazmatskog i izvanstaničnog dijela APP, te rasprostranjenost u gotovo svim tkivima ukazuje da APP ima i druge funkcije, od kojih su najistraženije one u proliferaciji stanica, zacjeljivanju rana i smanjivanju staničnog stresa. Točnije poznavanje funkcije APP i regulacije ekspresije njegovog gena moglo bi pomoći davanju odgovora na pitanje zašto samo u određenim populacijama neurona, te u samo nekim dijelovima živčanog sustava dolazi do odlaganja amiloida. Kako Ab nastaje u većini tjelesnih stanica, još uvijek nije jasno da li njegova prekomjerna količina primarno nastaje u mozgu ili izvankranijalno (pa u mozak dolazi hematogeno). Jedna od važnih, još uvijek nepotvrđenih hipoteza, je da nakon traume glave dolazi do poremetnje krvno-moždane barijere uslijed čega se cirkulirajući AbPP odnosno Ab prekomjerno nakuplja u mozgu.
APP ima kratko poluvrijeme života jer se brzo katabolizira, a ovaj događaj je poremećen u osoba s mutacijom APP, PS-1 i PS-2 gena. U katabolizmu AbPP sudjeluju tri enzima, nazvana a, b i g sekretaza (Shema 1). a-sekretaza cijepa AbPP između lizina na 687. i leucina na 688. poziciji, a zatim slijedi cijepanje g-sekretazom na poziciji 711 ili 713 (Shema 1). Budući da a-sekretaza cijepa AbPP oko sredine Ab domene, ovaj put ne dovodi do nastanka Ab (stoga je nazivan a, sekretorni ili fiziološki put), već nastaju N-terminalni fragment - topljivi AbPPa (sAbPPa, od ‘soluble AbPPa’), koji je normalan sastojak svih tjelesnih tekućina s dokazanim zaštitnim djelovanjem, i C-terminalni membranski fragment (naziva se još i p3 peptid jer je težak 3kDa). Tzv. endosomalno-lizosomalni put cijepanja AbPP započinje b-sekretaza koja ga cijepa između 671 i 672 aminokiseline, a zatim djeluje još i g-sekretaza na poziciji 711 ili 713/714. Ovaj put dovodi do nastanka amiloidogenične molekule Ab čija dužina (Ab1-40 ili Ab1-42(43)) ovisi o poziciji cijepanja g-sekretazom. Dulji oblik (Ab1-42(43)) naročito je važan jer čini najveći dio NP, toksičniji je i skloniji stvaranju fibrila (osim ova dva oblika, plakovi sadrže i manje količine drugih, manjih fragmenata Ab). Hipotetski slijed događaja koji uključuje mutaciju APP, PS-1 i PS-2 gena, poremećenu proteolizu AbPP, povećanu proizvodnju Ab, njegovu agregaciju i nakupljanje u obliku DP, a na koje se sekundarno vežu i ApoE, a1-antikimotripsin, serumski amiloid P, interleukin-1, bazični fibroblastni činitelj rasta, a2-makroglobulin, protein povezan s lipoproteinima male gustoće, perlekan i tridesetak drugih sastojaka, dovodeći do aktivacije mikro- i astroglije, tj. upalnog odgovora, s posljedičnim oštećenjem aksona, poremećajem homeostaze kalcija i metabolizma neurona) nazvan je amiloidna kaskadna hipoteza Alzheimerove bolesti (treba imati na umu da se depoziti amiloida javljaju i u nizu drugih bolesti i bolesnih stanja kao što su Down sindrom, Creutzfeldt-Jacobova bolest, Gerstmann-Sträussler-Scheinker sindrom, dijabetes tip 2, obiteljska amiloidna polineuropatija, multipli mijelom, itd.).
Neka od ključnih zapažanja na kojima se temelji amiloidna hipoteza su slijedeća: svi bolesnici s AD imaju NP sačinjene od Ab čija je brojnost i količina veća od one u normalnog starenja; većina do sada poznatih rizičnih gena za nastanak AD (APP, PS-1, PS-2, APOE4) dovode do pojačane produkcije Ab; uslijed jedne suvišne kopije APP gena na trećem 21-om kromosomu, sve osobe s Down sindromom prije 50 godine života razviju neuropatološke promjene karakteristične za AD (najranija prisutnost izvanstaničnog Ab u moždanom tkivu djece s Down sindromom otkrivena je već u 21. tjednu trudnoće, a SP već u 11. godini života, dok se prve neurofibrilarne promjene mogu vidjeti tek desetak godina kasnije); transgenični miševi sa mutiranim AbPP genom čovjeka razviju najprije DP, a zatim i NP sačinjene od Ab, što dovode do aktivacije mikroglija stanica i degeneracije neurona. Da je metabolizam poremećen puno prije nego što dođe do nastanka kliničkih simptoma potvrđuje i činjenica da fibroblasti osoba s FAD (kod ‘švedske’ mutacije APP) luče i do 3 puta veću količinu Ab 15 godina prije nastupa simptoma bolesti. Ipak, iako je Ab nesumnjivo važan činitelj u AD, ima i kontradiktornih nalaza koje je teško objasniti. Kad se Ab, u in vivo eksperimentima, injicira u moždano tkivo ne izaziva nikakav toksičan učinak. Također, ako se pretpostavi da Ab ima štetno djelovanje ili difundira u okolinu SP tada bi se očekivao i gradijent oštećenja oko SP, kojega nema. Također je vrlo malo uvjerljivih dokaza o direktnoj povezanosti SP s ostalim neuropatološkim obilježjima AD i kliničkim simptomima. Jedan dio istraživača zato misli da je unutarstanični Ab možda važniji u patogenezi bolesti od izvanstaničnog.
Zasad je poznato da aktivnost a-sekretaze posjeduje nekoliko enzima tzv. ADAM obitelji metaloproteaza: TACE (od ‘tumor necrosis factor a converting enzyme’ ili ADAM-17), MDC9 i ADAM-10. b-sekretazu su u Golgi aparatu i endosomima 1999. godine otkrile istovremeno i neovisno četiri skupine istraživača, a nazvana je BACE (od ‘b-site APP cleaving enzyme’). No, do danas još uvijek nije sekvencionirana g-sekretaza, iako je nekoliko neovisnih skupina tijekom 2000. godine podastrlo indirektne dokaze da je vjerojatno sam PS-1 g-sekretaza. Trenutni problem sastoji se u činjenici da je g-sekretaza pronađena u velikom proteinskom kompleksu koji se sastoji od b-katenina, kalsenilina, nikastrina, PS-1 i još nekoliko drugih s presenilinom povezanih proteina. Zna se da je g-sekretazna aktivnost posredovana kroz početni stadij vezanja, te naknadno djelovanje na proteolitičkom mjestu, a pretpostavlja se da su za to dovoljni samo nikastrin i PS-1.
Agregacija Ab ovisna je o njegovoj koncentraciji, pH i duljini inkubacije. Vodena otopina Ab pokazuje više kinetičku nego termodinamičku topljivost, tj. nakon dovoljno dugog razdoblja u njoj Ab može precipitirati. Različiti biometali induciraju agregaciju Ab in vitro, npr. bakar i željezo, a naročito cink (u jednom je istraživanju 1991. godine peroralno davanje cinka značajno i brzo dovelo do pogoršanja demencije u bolesnika s AD). Pored ovih elemenata, u dementnih je bolesnika opisan i poremećen metabolizam kadmija, mangana, žive, selena i radonovih 'kćeri', tj. alfa čestica polonija i beta čestica bizmuta. Budući da nakon različitih obliak staničnog stresa depolarizacija neurona dovodi do masivnog otpuštanja cinka u izvanstaničnu tekućinu, pretpostavlja se da povišene koncentracije cinka (i bakra) pospješuju agregaciju Ab i in vivo (a pri tome ApoE4 jače od ApoE3 potpomaže Ab izazvanu porastom koncentracija ovih metala). Neki istraživači prepostavljaju da je upravo jedna od najvažnijih zadaća APP transport iona bakra i cinka. U postmortalnom moždanom tkivu AD bolesnika pronađena je i značajna količina aluminija, kako u središnjim dijelovima SP tako i u neuronima s NFT, a injekcija aluminijevih soli u mozak zeca dovodi do nastanka NFT. Neke epidemiološke studije utvrdile su povezanost razine aluminija u pitkoj vodi i učestalosti AD, a davanje helata koji vežu aluminij u nekim je studijama usporilo progresiju AD. Ipak, većina istraživanja nisu potvrdila navedenu povezanost između aluminija i AD.
Vjerojatno najbolje upoznato neurotoksično djelovanje aluminija je ono u bolesnika na dugotrajnoj hemodijalizi. Prepoznavanje ovog poremećaja dovelo je do strogih kriterija čistoće vode za dijalizu, jer su mnogi od ovih bolesnika postali dementni. Dijalitičku demenciju najčešće karakteriziraju poteškoće u govoru, mioklonus i epilepsija. Prestanak korištenja vode zagađene aluminijevim hidroksidom prilikom hemodijalize dovodi do djelomičnog nestanka simptoma. Neki od ovih bolesnika imaju depozite Ab u obliku DP, ali bez NP i NFT. Iako brojnost DP prelazi onu koja se susreće u normalnom starenju, njihov utjecaj na kliničku sliku nije jasan. Kao i u slučaju akutne traume glave, odlaganje Ab i ovdje može predstavljati samo ‘odgovor na stres’, a povišena razina aluminija uzrokom kliničkih simptoma putem drugih mehanizama.
Procesi normalne razgradnje i odstranjivanja Ab nisu dobro poznati, a također mogu predstavljati kritična točke za nastanak AD. Ab najbolje razgrađuje katepsin D pri pH4-5. Putem otpuštanja proteaza disintegrinske obitelji i mikroglija može utjecati na razgradnju Ab. a2-makroglobulin pospješuje odstranjivanje Ab putem LRP receptora.
Do sada je otkriveno više od 10 mutacija APP gena (najčešće mutacije su: N665D, otkrivena 1994., početak bolesti nastupa u prosjeku s 86 godina; K/M670/671N/L, otkrivena 1992., početak bolesti nastupa u prosjeku s 52 godine (44-59), tzv. švedska mutacija; A692G, 1992., početak s 40-60 g., tzv. flamanska mutacija; E693G, 2000., početak s 58 g., tzv. arktička mutacija; E693Q, 1990., tzv. nizozemska mutacija – uzrokuje HCHWA-D; E693G, dob početka 58.g.; A713V, 1992., dob početka 78.g.; T714I, 2000., dob početka 40 godina, smrt obično u roku od 2 godinem, tzv. austrijska mutacija; V715M, 1999., dob početka 52 g. (40-60), tzv. francuska mutacija; I716V, 1997., dob početka 55 godina, tzv. floridska mutacija; V717I, 1991, dob početka 55 godina (50-60 god.), tzv. londonska mutacija; V717F, 1991., dob 47 (42-52); V717G, 1991., dob 55 (50-60); V717L, 2000., dob početka kasne tridesete, brza smrt; L723P, 2000., australijska mutacija), te više od 70 mutacija PS-1 gena i 5 mutacija PS-2 gena. Sve su ove mutacije povezane s akumulacijom dužeg oblika Ab, dok je povećana količina kraćeg oblika nađena samo u Down sindromu i tzv. švedskoj mutaciji AbPP. Vaskularni amiloid sačinjen je samo od kraćeg oblika Ab. DP inicijalno sadrže samo dulji, na koji se kasnije postepeno deponira i kraći oblik Ab. Samo jedna od do sada otkrivenih mutacija APP gena ne dovodi do AD, već do nasljednog cerebralnog krvarenja s amiloidozom - nizozemski tip (HCHWA-D, od ‘hereditary cerebral hemorrhage with amyloidosis - Dutch type’). U ovom poremećaju ne dolazi do nastanka NFT, a od SP se vide samo rani stadiji, jer amiloid nakupljen u mediji moždanih krvnih žila relativno rano (u pedesetim) uzrokuje smrtonosne infarkte. Budući da je ovdje vaskularni sustav izvor Ab, HCHWA-D podupire hipotezu o hematogenom nastanku AD. Neki istraživači misle da je AD sistemska autoimuna bolest s cirkulirajućim imunokompleksima što učestvuju u nakupljanju Ab u moždanom tkivu. Ovi imunokompleksi, osim Ab, sadrže i IgG i IgM protutijela, te C3 komponentu komplementa, a čini se da ih ima statistički značajno više u koži bolesnika s AD nego u kontrola.
Još uvijek nije sasvim jasno kako apolipoprotein E4 utječe na nastanak i progresiju AD, ali je poznato da Ab potiče ekspresiju apoE u astrocitima, da izlučeni apoE smanjuje upalnu reakciju, tj. aktivaciju mikroglije in vitro, te da apoE3 i apoE2 stvaraju stabilne komplekse s Ab (koji se onda možda odstranjuju putem LDL receptora). Zato se misli da prisutnost apoE4 povećava vjerojatnost agregacije i taloženja Ab i izazivanja upalnog odgovora. Osim toga, čini se da apoE4 više od apoE3 i apoE2 potiče endosomalno-lizosomalni proteolitički put APP, te ubrzava stvaranje konformacije beta uzvojnice Ab. Prema drugoj pretpostavci, apoE4 onemogućuje tau proteinima stabilizaciju mikrotubula, dovodeći do nastanka NFT. U svakom slučaju, daljnje je proučavanje uloge apoE izvanredno važno, jer bi spojevi koji oponašaju djelovanje apoE2 (pod uvjetom netoksičnosti i prolaska kroz krvno-moždanu barijeru) mogli biti korisni i u prevenciji i u liječenju AD.
2.6.2. Neurofibrilarna degeneracija i hipoteza poremećaja citoskeleta
Neurofibrilarna degeneracija je, iako nespecifično, još uvijek najkarakterističnije patološko oštećenje u AD. Ova strukturna abnormalnost očituje se neurofibrilarnim snopićima (NFT, od ‘neurofibrillary tangles’), vlakancima u neuropilu (NT, od ‘neuropil threads’) i distrofičkim neuritima (DN) SP (kao što je već spomenuto, SP koji sadrže ovakve neurite nazivaju se neuritičkim plakovima, NP).
Svjetlosno-mikroskopski, neurofibrilarna degeneracija nastaje pojavom zadebljanih, vijugavih vlakanaca unutar citoplazme većih neurona koja se grupiraju u NFT. Izgled NFT može varirati s obzirom na smještaj unutar citoplazme i tip neurona. Veliki kortikalni piramidni neuroni najčešće sadrže NFT u obliku plamička ('flame-shaped'), trokuta ili petlje, dok se štapićasti i klupkasti oblici češće vide u hipokampusu. U ‘dubokim’ jezgrama mozga, npr. bazalnoj, crnoj i raphe jezgrama, lokus ceruleusu i donjem dijelu moždanog debla, NFT imaju okruglast oblik (globozni NFT). S obzirom na nastanak, količinu i maturaciju nakupljenih vlakanaca, NFT se obično dijele na rane, zrele i izvanstanične. Izvanstanični NFT (‘ghost tangle', 'tombstone') nastaju kad neuron umre, a na njegovom mjestu ostane u neuropilu samo NFT i posljedična glioza, što je naročito izraženo u teškim i dugotrajnim slučajevima AD. Osim što se dobro prikazuju metodama srebrne impregnacije (na parafinskim rezovima obično pomoću Bielschowsky, Cross, Gallyas ili Palmgren metode, a na smrznutim rezovima von Braunmühlovom metodom), zbog fizikalnog svojstva da dvostruko lome svjetlo NFT se izvrsno prikazuju i nakon bojanja Kongo crvenilom osvjetljavanjem polariziranim svjetlom, te nakon bojanja tioflavinom S osvjetljavanjem ultraljubičastim svjetlom.
Pored toga što se mogu naći u starenju i AD, NFT se susreću i u značajnom broju drugih neuroloških bolesti i stanja. To su: progresivna supranuklearna kljenut, pugilistička (‘boksačka') demencija, Pickova bolest, frontotemporalna demencija s parkinsonizmom, bolest argirofilnih zrnaca, kortikobazalna degeneracija, subakutni sklerozirajući panencefalitis (NFT su u ovoj bolesti nađeni čak u 16-mjesečnog djeteta), Niemann-Pickova bolest tip C, trovanje olovom, amiotrofična lateralna skleroza, Guam demencija s parkinsonizmom, Down sindrom, postencefalitički parkinsonizam, miotona distrofija, kongenitalna mišićna distrofija (Fukuyama tip), Kufsova bolest, Cockayneov sindrom, Hallervorden-Spatzova bolest, Gerstmann-Sträussler-Scheinkerov sindrom, demencija neurofibrilarnih snopića s kalcifikacijom. Ponekad se NFT mogu vidjeti i u području arteriovenskih malformacija, kod mentalno retardiranih odraslih osoba, te u lipofuscinozi. Stoga se može zaključiti da nastanak NFT predstavlja zajednički završni put degeneracije neurona koji može nastati uslijed različitih virusnih, genetskih, metaboličkih i fizičkih poremećaja.
Elektronsko-mikroskopski, NFT se u AD sastoje od dvije dugačke, nerazgranate, integralne podjedinice (filamenta) sparene u dvostruku uzvojnicu (PHF, od ‘paired helical filament’). Obje podjedinice zajedno imaju promjer 20-22 nm, a uzvojnica se sužava u pravilnim razmacima svakih 80 nm. Svaka podjedinica sastoji se od najmanje 4 protofilamenta. Iako se većina NFT nađenih u AD i starenju sastoji prvenstveno od PHF, NFT mogu biti sačinjeni i od mješavine PHF i ravnih filamenata promjera 15-16 nm (SF, od ‘straight filament’), ili od isključivo jednih ili drugih. I PHF i SF se sastoje od tau proteina, ali su im podjedinice različito raspoređene. Koliko su međusobno slični najbolje govori činjenica da se često može vidjeti kako isti filament na jednom kraju ima izgled PHF, a na drugom kraju izgled SF (hibridni filamenti). NFT u PSP sastoje se prvenstveno od SF, a samo manjim dijelom od mješavine SF i PHF (svjetlosno-mikroskopski nije ih moguće razlikovati od NFT u AD). PHF i SF se također mogu naći i u astrocitima, ali samo u uznapredovalim, dugotrajnim slučajevima AD.
Vijugava vlakanca u međustaničnoj tvari (NT, od ‘neuropil threads’) dobro se prikazuju metodama srebrne impregnacije i tioflavinom, a predstavljaju 'treću lokaciju' (pored NFT i NP) PHF u AD. Svjetlosno-mikroskopski se vidi da NT predstavljaju degenerirane dendrite, a oko 10% ih čine aberantne aksonske grane. NT se javljaju vrlo rano u tijeku AD, a naročito ih puno ima u hipokampusu i entorinalnom korteksu. U prisutnosti NFT njihov broj značajno raste i pozitivno korelira s težinom bolesti. U normalnom starenju je pojavljivanje NT strogo ograničeno na entorinalni korteks, hipokampus i amigdala. NT i DN NP elektronsko-mikroskopski imaju veći omjer SF/PHF nego NFT, jednako kao i fibrilarni snopići u oligodendroglija stanicama. Gotovo 70% NT sadrži samo SF ili PHF, bez imalo normalnih filamenata. Kad se sve vrste filamenata prikažu kao proporcija ukupne količine filamenata u NT, uočava se obrnuta linearna korelacija između normalnih neurofilamenata i SF, te između SF i PHF. Stoga se pretpostavlja da i u aksonima i u dendritima zahvaćenim patološkim procesom u AD normalne neurofilamente postepenom transformacijom ili korištenjem nekih njihovih dijelova zamjenjuju SF, a oni se zatim postupno pretvaraju u PHF. Moguće je da hiperfosforilacija neurofilamenata prethodi hiperfosforilaciji tau proteina.
PHF se sastoje od hiperfosforiliranog proteina tau (t) koji polimerizacijom stvara filamente, što je dokazano in vitro, elektroforetski, imunocitokemijski, elektronsko-mikroskopski, sekvencioniranjem i masenom spektroskopijom. No, abnormalni tau može biti prisutan u stanicama i u nepolimerizirnom obliku. Pored tau proteina, u PHF se u maloj količini sekundarno nađe i ubikvitin (koji ‘obilježava’ abnormalne proteine za razgradnju nelizosomalnim proteazama), kao i brojni drugi sastojci.
Tau protein spada u tzv. drugu skupinu s mikrotubulima povezanih proteina (MAP, od ‘microtubule associated proteins’) karakteriziranu s 3 ili 4 ponavljajuća slijeda od 18 aminokiselina, koji služe povezivanju s mikrotubulima (MT). Tau protein čini tzv. kratke, 18 nm dugačke ‘ručice’ što premošćuju MT u aksonima (dugačke mostove čini MAP1B). Kada ovi MAP u potpunosti okruže MT, podjedinice a i b tubulina u MT ne mogu se razdvojiti.
Tau protein kodira jedan gen dugačak oko 100 kb smješten na dugom kraku kromosoma 17 (17q21). Aktivnost promotora prepisivanja tau gena značajno se povećava za vrijeme inicijacije nastanka i izrastanja aksona. Primarni transkript tau gena sastoji se od 13 eksona, od kojih se eksoni –1 i 14 ne translatiraju. Eksoni 1, 4, 5, 7, 9, 11, 12 i 13 su konstitutivni, a naizmjeničnim sparivanjem eksona 2, 3 i 10 nastaje u odraslih osoba obitelj od 6 izo-oblika. Jedinstveni obrazac ekspresije izo-oblika tau proteina u mozgu čovjeka ukazuje na njegovu moguću povezanost s izraženom sklonošću čovjeka prema neurodegenerativnim bolestima s tauopatijom, naročito AD. Budući da se različito eksprimiraju za vrijeme razvitka, pretpostavlja se da svaki pojedini izo-oblik ima posebnu regulaciju i fiziološku ulogu. U mozgu fetusa eksprimira se samo jedan izo-oblik (fetalni tau), kojemu nedostaju dijelovi što ih kodiraju eksoni 2, 3 i 10 i stoga je najkraći oblik od 352 aminokiseline (najdulji oblik ima 441). Adultni oblici tau proteina pojavljuju se tek postnatalno, a različito se eksprimiraju u različitim populacijama neurona. Tako npr. glasnička RNA za tau u zrnatim stanicama fascije dentate ne sadrži ekson 10, što je možda razlogom zašto ove stanice gotovo nikad ne razviju NFT, čak ni u najtežim i najdugotrajnijim slučajevima AD. U većini normalnih neurona lokalizacija tau proteina ograničena je na aksone, a samo u manjoj mjeri se on nalazi i u dendritima, te u glija stanicama. Tau protein u gliji razlikuje se od neuronalnog po tome što mu nedostaje segment kodiran eksonom 3.
Tau protein sastoji se od projekcijske domene i domene što služi povezivanju s MT. Domena za povezivanje s MT kopolimerizira s tubulinom, a stimuliranjem vezanja GTP za b podjedinicu tubulina, regulira i brzinu međusobnog spajanja MT. Sastoji se od 4 visoko konzervirana ponavljajuća slijeda R1-R4 što se sastoje od po 31 ili 32 aminokiseline, a kodiraju ih eksoni 9-12. Odrasli tau izo-oblici s 4R (R1-R4) su oko 40 puta učinkovitiji u promociji polimerizacije MT nego fetalni tau kojem nedostaje ekson 10 (pa stoga ima 3R: R1, R3 i R4). Čini se da je izostanak ekspresije R2 za vrijeme fetalnog razvitka neophodan za visoku plastičnost citoskeleta rastućih aksona nezrelih neurona. Projekcijska domena tau proteina veže se za neurofilamente ili druge MT, što ukazuje da je funkcija tau i integracija MT s ostalim elementima citoskeleta neurona. Putem SH3 domena src kinaza, fosfolipaze Cg, spektrina i aktina, tau je u stalnoj interakciji s događajima na membrani neurona.
Biološka aktivnost tau proteina kontrolirana je tijekom razvitka fosforilacijom. Stupanj fosforilacije visok je u fetalno doba, a postnatalno sve više slabi zbog aktivacije fosfataza. Poremećena fosforilacija svih šest izo-oblika tau proteina u odraslom mozgu mijenja njegovu konformaciju dovodeći do disocijacije tau proteina od MT. Slijedi progresivna ubikvitinacija i digestija tau proteina, te nastanak visoko netopljivih PHF. Za netopljivost PHF nije odgovorna hiperfosforilacija, već unakrsno povezivanje tau proteina proteina putem tkivne transglutaminaze. Abnormalni tau proteini u AD sadrže i do četiri puta više fosfata po molu proteina (normalni imaju 2.5-3.5, a abnormalni i do 10-12 mola fosfata po molu proteina, obično oko 6-8), jer su fosforilirani na više mjesta. Imunocitokemijski i masenom spektroskopijom dokazano je da je tau najčešće fosforiliran u PHF u AD na slijedećih devet mjesta: Ser-46, Thr-123, Ser-199, Ser-202, Thr-231, Ser-235, Ser-262, Ser-396 i Ser-404 (oznake prema najduljem izo-obliku). Od sveukupno 80 mogućih mjesta fosforilacije, do danas ih je dokazano više od 30, ali ona nisu nikad istovremeno fosforilirana (to bi iznosilo više od 20 mola fosfata po molu proteina). Razlog za to je vjerojatno činjenica da se PHF kontinuirano fosforiliraju i defosforiliraju, a hiperfosforilacija već i relativno manjeg broja mjesta dovodi do konformacijskih promjena i nastanka PHF. Za stanje povećane fosforiliranosti tau proteina može biti odgovorna aktivnost najmanje 15 različitih kinaza (glikogen sintaza kinaza-3, kalcij/kalmodulin ovisne protein kinaze, stresom i mitogenima aktivirane protein kinaze, o ciklinu ovisne protein kinaze, kazein kinaza II, fosforilaza K, kinaza ovisna o cAMP (PK-A), itd.), a s druge strane, i nedostatak ili smanjena aktivnost fosfataza može dovesti do istog učinka. Tako su u AD opisane smanjene razine fosfataza 1, 2A, 2B i 2C.
Hiperfosforilacija tau proteina vjerojatno doprinosi i početku i progresiji AD, a regionalne razlike u metabolizmu tau proteina možda predstavljaju molekularni temelj za individualno i regionalno različitu vulnerabilnost neurona u AD. Broj hiperfosforiliranih tau-reaktivnih neurona gornje vijuge sljepoočnog režnja pokazuje jaku pozitivnu korelaciju sa stupnjem demencije i gubitkom sinapsi čeonog režnja. Ovaj je nalaz vjerojatno posljedica činjenice da se imunološka reakcija na fosforilirane epitope tau proteina odvija i prema neuronima čije je djelovanje već poremećeno iako nisu stigli oblikovati PHF. Čini se da je od prvih blago promijenjenih neurona uslijed lakše hiperfosforilacije, pa do nastanka izraženih neurofibrilarnih promjena i simptoma demencije u sporadičnih slučajeva AD potrebno najmanje oko 20-30 godina, jer se prvi takvi neuroni mogu primijetiti tek u osoba starijih od 30 godina. Vjerojatno uslijed nemogućnosti ispunjavanja stalnih zahtjeva za plastičnošću ključnih dijelova mozga (tzv. hipoteza sloma neuronalne plastičnosti) povezanih s učenjem i pamćenjem (entorinalni korteks, hipokampus, bazalni telencefalon), u bolesnika s AD također je uočena povećana ekspresija fetalnog tau, kao i ponovna aktivacija (zasad) jedne fetalne kinaze koja fosforilira tau.
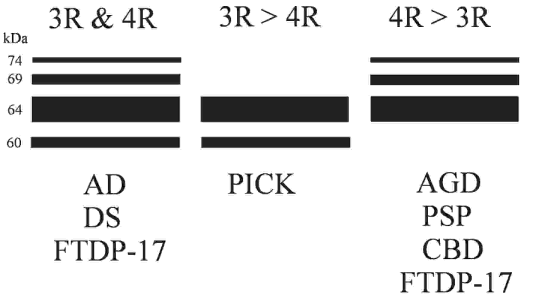
Hiperfosforilirani tau proteini akumuliraju se u neuronima i glija stanicama
na način koji je specifičan za odredenu neurodegenerativnu bolest. Tako se 3R
i 4R izo-oblici nalaze u različitim proporcijama u AD, Down sindromu i nekim
obiteljima s frontotemporalnom demencijom s parkinsonizmom uslijed mutacija
tau gena (FTDP-17), 3R oblici prevladavaju u Pickovoj bolesti, dok 4R oblici
prevladavaju u progresivnoj supranuklearnoj kljenuti (progressive supranuclear
palsy, PSP), kortikobazalnoj degeneraciji (CBD), nekim oblicima FTDP-17 i bolesti
argirofilnih zrnaca (Braakovoj demenciji) (vide infra) (Shema 2). Trenutno se
pokušavaju usavršiti postupci analize cerebrospinalne tekućine kojima bi se
iz likvora finom analizom tau proteina mogla točno utvrditi određena neurodegenerativna
bolest. Od posttranslacijskih promjena, osim fosforilacije, tau proteini podložni
su i O-glikozilaciji (tj. dodavanju N-acetil-glukozamina na atom kisika) na
serine i treonine koji se nalaze u blizini prolina.
Sve do posljednjih nekoliko godina amiloidna kaskadna hipoteza dominirala je
razmišljanjem većine znanstvenika koji se bave AD. Medutim, brojni novi podaci
ukazuju da poremećaji citoskeleta također imaju ogromnu važnost u nastanku AD.
Prema ovoj koncepciji amiloid ima relativno malu, perifernu ulogu u patogenezi
AD, dok destabilizacija citoskeleta nije samo puko nakupljanje PHF, vec ključni
činitelj za nastanak svih ostalih patoloških promjena. U prilog ovoj tvrdnji
govori i činjenica da sva četiri dokazana rizična gena za nastanak AD (APP,
PS-1, PS-2 i APOE) na ovaj ili onaj način remete citoskelet neurona, s posljedicnim
raspadom Golgi aparata, poremećenim procesiranjem proteina i aksonskim transportom.
Rezultat je gubitak sinapsi i neurona što dovodi do diskonekcijskog sindroma
i demencije. Uloga s mikrotubulima povezanih kinaza i fosfataza u nastanku neurofibrilarnih
promjena još uvijek nije razjašnjena, ali ima velik potencijalni značaj jer
bi se njihovom manipulacijom moglo zaustaviti stvaranje PHF kako u AD, tako
i u 'normalnom' starenju.
Shema 2. Elektroforetski profil tau proteina u različitim neurodegenerativnim
bolestima (za objašnjenje vidi tekst).
2.6.3. Neuropatološka dijagnoza
Iako je AD primarno bolest moždane kore, ona ne predstavlja generalizirani gubitak kortikalnih funkcija već lokalizacija promjena pokazuje specifičan obrazac s obzirom na populacije neurona zahvaćenih patološkim procesom, tj. anatomski smještaj neurona s obzirom na područje i sloj korteksa, te neurotransmitersku narav. Arealna i laminarna distribucija NFT u neokorteksu jasno ukazuje da su u tijeku AD najranije i najjače zahvaćene populacije neurona veliki piramidni, glutamatergički neuroni sloja V čiji aksoni čine duge kortikokortikalne projekcije, a zatim i piramidni neuroni dubokog sloja III. Najviše su zahvaćeni sljepoočni, a zatim čeoni i tjemeni režnjevi. Manji piramidni neuroni slojeva II, VI, i gornjih dijelova sloja III otporniji su na nastanak NFT, a zvjezdoliki neuroni i mali piramidni neuroni sloja IV, kao i sve vrste inhibitornih neurona, izvanredno su otporni na nastanak NFT. Činjenica da sloj V pokazuje veću gustoću NFT nego površni slojevi asocijativnih područja korteksa ukazuje da su povratne (započinju u dubokim slojevima korteksa i završavaju u prvom i šestom sloju) i lateralne projekcije (započinju u dubokim slojevima, a projiciraju se u sve slojeve) jače zahvaćene nego unaprijedne (započinju u površnim slojevima, a završavaju u četvrtom i dubokom trećem sloju ciljne areje). SP se obično nalaze u međustaničnoj tvari na mjestima gdje završavaju kortikokortikalne i kortikofugalne projekcije neurona zahvaćenih s NFT (to su opet većinom asocijativna područja), što upućuje da su u nastanak SP uključeni brojni neuronski sustavi.
Navedena načela vrijede i za hipokampalnu formaciju, pa tako eferentni neuroni sloja II entorinalnog korteksa iz kojih započinje perforantni put pokazuju najraniju i najjaču zahvaćenost patološkim promjenama u tijeku AD, a slijede piramidni neuroni sloja V entorinalnog korteksa, te veliki piramidni neuroni subikuluma i CA1 područja. Prisutnost završnih stadija NFT u ovim neuronima dobro korelira s prisutnošću NP na mjestu završetka njihovih vlakana. Tako je nastanak NFT u neuronima II sloja entorinalnog korteksa povezan s nastankom NP u molekularnom sloju girusa dentatusa, a slično se visoka učestalost NFT u sloju V entorinalnog korteksa može povezati sa degeneracijom veza i nastankom NP u specifičnim jezgrama amigdala i drugim limbičkim i asocijativnim kortikalnim područjima. Iako ima izuzetaka koji se ne mogu objasniti na ovaj način, čini se da je suprotno navedenoj djelomičnoj povezanosti NFT s NP, povezanost između DP i stvaranja NFT vrlo slaba. Tako se nakupljanje Ab najprije vidi u sljepoočnoj moždanoj kori, a ne u entorinalnom korteksu ili u hipokampalnoj formaciji, bez obzira na neuropsihološki status pacijenta. Stoga se može reći da demencija prvenstveno odražava gubitak funkcije skupina visokospecijaliziranih kortikalnih neurona, a ne korelira s odlaganjem amiloida. Demencija i gubitak neurona u pozitivnoj su korelaciji s brojem NFT u asocijativnim područjima moždane kore, dok broj SP ne korelira ni s kognitivnim oštećenjem niti s gubitkom neurona. Neke skupine istraživača ipak još uvijek pokušavaju dokazati da se takva korelacija ipak može ustanoviti s obzirom na ukupnu količinu amiloida (a ne broj SP).
Tradicionalno se završna neuropatološka dijagnoza AD donosi na temelju kliničko-patološke korelacije, kao što je to činio i sam Alzheimer. Od predloženog niza neuropatoloških kriterija za donošenje dijagnoze AD, najkorišteniji je dugo godina bio bio NIA (od ‘National Institute of Aging’) kriterij. NIA kriterij se sastoji od brojanja SP, a dobiveni broj se prilagođuje starosti osobe. Tako je minimalni broj SP, za dijagnozu AD u pacijenta starosti 50-65 godina iznosio 8 ili više po mm2, 66-75 godina 10 ili više, te za osobe starije od 75 godina 15 ili više po mm2. NIA kriterij ne određuje vrstu SP (što je loše jer se u AD, za razliku od normalnog starenja, karakteristično nalazi veći broj NP, nego DP, pogotovo u mlađih bolesnika), pa je preosjetljiv (dio normalnih nedementnih starijih osoba ima dovoljno SP za dijagnozu AD). Ovaj kriterij također nije određivao ni područje neokorteksa u kojem bi se kvantifikacija trebala izvršiti (zato mu je bila mala specifičnost, a rezultati nestalni i neprimjereni za usporedbu između ustanova). Iz navedenih je razloga 1993. godine ustanovljen CERAD (od ‘Consortium to Establish a Registry for Alzheimer’s Disease’) kriterij. CERAD kriterij se temelji na polukvantitativnoj analizi NP pomoću modificiranog Bielschowsky ili tioflavin S bojanja u tri definirana područja moždane kore (gornja sljepoočna vijuga, prefrontalni korteks i donji tjemeni režnjić), a također je usklađen se starošću ispitanikova mozga. U kombinaciji s kliničkim pokazateljima demencije, na temelju broja NP donosi se dijagnoza moguće, vjerojatne ili definitivne AD. Kako neokortikalni NFT nisu uvjet za dijagnozu AD prema CERAD kriteriju (što je loše jer se, za razliku od ‘normalnog’ starenja u kojem se NFT javljaju samo u sljepoočnom režnju, u većini AD slučajeva nađu neokortikalni NFT), kao ni patološke promjene hipokampusa, entorinalnog korteksa i amigdala, niti ovaj kriterij nije idealan. Stroži kriteriji od CERAD-a (NIA-RI, od ‘National Institutes of Aging – Reagan Institute’, 1997) ipak se nisu pokazali značajno boljima, jer su uzimajući u obzir broj neokortikalnih NFT postali previše specifični i nedovoljno osjetljivi, budući da značajan broj dementnih bolesnika s AD ima mali broj neokortikalnih NFT. Najveći problem svih navedenih kriterija je činjenica da broj SP ne korelira dobro sa stupnjem kognitivnog oštećenja, tj. demencijom.
Kako najveću pozitivnu korelaciju između neuropatoloških promjena i stupnja demencije daje upravo broj NFT, Braak i Braak od 1991. godine predlažu novu shemu analize i donošenja neuropatološke dijagnoze na temelju broja NFT u entorinalnom korteksu, hipokampusu, asocijativnom sljepoočnom korteksu, te primarnom i asocijativnom vidnom korteksu. S obzirom na nastanak NFT i progresivne promjene hipokampalne formacije oni su tijek AD podijelili u 6 neuropatoloških stadija (I-VI), a s obzirom na odlaganje Ab u 3 stadija (A-C). Najraniji stadiji pokazuju blagu, ali značajnu zahvaćenost transentorinalnog (karakteristično se NFT postepeno spuštaju od drugog prema dubokom dijelu trećeg sloja) i entorinalnog korteksa sa NFT (stadiji I i II), te odlaganje Ab u bazalnim dijelovima sljepoočne moždane kore (stadij A). Ovi su dijelovi moždane kore selektivno zahvaćeni patološkim procesom još u vrijeme kad nema kliničkih znakova demencije koji bi se mogli otkriti neuropsihološkim testiranjem. U stadijima III i IV, NFT se prošire i na ostala limbička područja, a količina Ab se postupno povećava u limbičkom režnju i neokortikalnim područjima (stadij B). Patološke promjene u ovom stadiju dovode do prekida veza unutrašnjih veza hipokampalne formacije, kao i između hipokampalne formacije i neokortikalnih područja. Tako hipokampalna formacija ostaje izolirano područje, s oštećenim projekcijama u i iz drugih limbičkih i neokortikalnih područja (diskonekcijski sindrom). Ove patološke promjene klinički odgovaraju ranom stadiju AD. U završnim stadijima V i VI gotovo cijeli izokorteks bude zahvaćen s NFT, a depoziti Ab mogu se naći i u primarnim kortikalnim područjima (stadij C). Klinički, ove promjene odgovaraju stadiju potpuno razvijene bolesti. Nažalost, zbog metodološke zahtjevnosti ovaj kriterij se ne koristi u svakodnevnoj praksi, a i NIA-RI Radna grupa predlaže ga još uvijek samo za znanstvena istraživanja. Važnost određivanja Braakovih stadija ogleda se u činjenici da je blaga do jača zahvaćenost entorinalnog korteksa i CA1 područja hipokampusa podudarna s normalnim do blago poremećenim kognitivnim statusom (kakav se često susreće i u nedementnih starijih osoba) još uvijek dovoljnim za svakodnevni život bez pomoći drugih, dok je širenje degenerativnih promjena na neokortikalna područja, s posljedičnom značajnom degeneracijom kortikokortikalnih veza, povezano s izraženim kliničkim znakovima demencije.
2.6.4. Ostala histopatološka obilježja i uloga glija stanica
Granulovakuolarna degeneracija (GVD) lako se uočava već i pomoću hematoksilin-eozinskog ili srebrnih bojanja, a najprisutnija je u CA1 području hipokampusa i subikulumu gdje se vakuole nalaze u citoplazmi piramidnih stanica. GVD se pojavljuje uglavnom u neuronima sklonim nastanku NFT, a ponekad se može naći i u supstanciji inominati i amigdaloidnom kompleksu jezgara. Pojedinačna vakuola ima promjer 3-4 mikrometra, a u sredini sadrži gusto zrnce promjera 1-2 mikrometra. Stanice koje pokazuju GVD relativno su rijetke u tijeku normalnog starenja, dok ih u AD ima puno. Pokazuju imunoreaktivnost na tau, tubulin i neurofilamente.
Hiranova tjelešca (HB) također se najčešće nalaze u piramidnim stanicama hipokampusa. Najprije su opisana kod bolesnika s Guam demencijom s parkinsonizmom, a tek su kasnije uočena i u AD. To su eozinofilični štapići koji se nalaze, kao i GVD, gotovo isključivo u ili uz piramidne stanice hipokampusa, a debeli su 7 do 10 mikrometara. Elektronsko-mikroskopski se sastoje od alternirajućih redova filamenata, a imunocitokemijski pokazuju reaktivnost na aktin, tropomiozin i druge bjelančevine mišića. Nisu specifična za AD, jer se mogu naći i u normalnih starijih osoba.
Za amiloidnu angiopatiju (AA) najvažnije je reći da ona obično zahvaća leptomeningealne krvne žile srednje veličine, i to najviše u zatiljnim (naročito bogati vaskularni pleksus sloja IVc primarnog vidnog korteksa) i cerebelarnim područjima, dok je hipokampalno područje pošteđeno. Ovakva je raspodjela zanimljiva jer je suprotna raspodjeli SP i NFT. Najraniji infiltrati amiloida u krvnim žilama vide se kao kongofilna ili tioflavin pozitivna tvar, imaju oblik finih filamenata promjera 80 nanometara i nalaze se u bazalnoj membrani. Kako infiltracija napreduje tako ovi filamenti zadebljavaju, te zahvaćaju i mediju i adventiciju. Amiloid obično zahvaća samo jedan dio cirkumferencije krvnih žila, a usprkos ovako promijenjenoj stijenci krvnih žila, krvarenja su u AD rijetka. Ispod bazalne membrane kapilarnog zida može doći do infiltracije amiloida u susjedni parenhim.
Budući da se aktivirana mikroglija nalazi u i oko SP, pretpostavljeno je da aktivacija imunološkog sustava ima ulogu u nastanku SP i patogenezi AD. Za razliku od rezidentne mikroglije koja ‘miruje’ u neuropilu, aktivirane su mikroglija stanice hipertrofične i imaju debele i ramificirane produljke, te visoku ekspresiju antigena klase II glavnog sustava tkivne histokompatibilnosti (HLA MHC-DR, od ‘human leukocyte antigen major histocompatibility complex’), a nešto manje i klase I, te C1q i C3 komponenata sustava komplementa, interleukina-1a (IL-1a), feritina i drugih. Jedna od mnogobrojnih hipoteza pretpostavlja da prekomjerno odlaganje Ab inducira aktivaciju mikroglije, koja zbog toga dobiva fagocitička svojstva, te luči IL-1 i druge citokine, koji dovode do pretvorbe SP u NP. Tretiranje mikroglija stanica u kulturi s Ab dovodi do njihove sekrecije činitelja nekroze tumora (TNFa, od ‘tumor necrosis factor’) i dušičnog monoksida, a budući da su obje ove molekule potencijalno neurotoksične, ovaj nalaz potvrđuje mguću degeneraciju neurona uslijed aktivacije mikroglije. No, i pored toga, ne može se isključiti mogućnost da su aktivirane mikroglija stanice samo kasna posljedica uznapredovalog patološkog procesa. Neočekivano, ali čini se da broj aktiviranih mikroglija stanica u hipokampalnoj formaciji bolje korelira s ukupnim brojem NFT u dotičnom području, nego sa SP, možda zbog povezanosti s izvanstaničnim NFT.
Jedno od glavnih svojstava astrocita je njihov jaki odgovor na široki spektar neuroloških oštećenja, a različite promjene koje čine ovaj odgovor jednim se imenom nazivaju astroglioza. Glavno obilježje ovog fenomena je povećana ekspresija glijalnog fibrilarnog kiselog proteina (glial fibrillary acidic protein, GFAP). Dobro je poznato da Ab inducira ovakav reaktivni fenotip astrocita u kulturi, no mehanizmi aktivacije astrocita u AD slabo su poznati. Najčešće je spominjana hipoteza aktivacija putem IL-1b i drugih citokina koje luče mikroglija stanice. Iz ovoga nalaza proizašla je hipoteza da AD možda predstavlja poremećaj gdje odgovor organizma značajno, a možda i esencijalno, utječe na nastanak patoloških promjena, dok inicijalni inzult (Ab) ima samo minorne posljedice. Vjeruje se da je uloga astrocita stvaranje zida prema mjestu oštećenja, prvenstveno onih izazvanih prevelikom količinom kalcija. Brojna istraživanja ukazuju da je jedan od najvažnijih činitelja koji određuje povezanost reaktivnih astrocita s plakovima razvojni stadij u kojem se plak nalazi. I DN i reaktivni astrociti mogu se vidjeti samo u primitivnom i neuritičkom stadiju, ali ne u početnom, stadiju difuznih depozita Ab. Stoga se čini da bi prisutnost reaktivnih astrocita na neki način doprinosila promjeni konformacijskog stanja Ab (i prijelaza plaka iz difuznog u primitivni oblik), što je potvrđeno in vitro. Ipak, nije jasno da li astrociti postaju reaktivni uslijed signala iz degeneriranih neuritičkih nastavaka ili pak oni šalju signale koji dovode do degeneracije neuritičkih nastavaka. Relativna proporcija plakova oko kojih se nalazi reaktivna astroglija u odsutnosti DN relativno je visoka u inicijalnim i blagim slučajevima AD, te značajno opada tijekom bolesti. Stoga se čini je s plakovima povezana astrocitoza vrlo rani (a stoga toga možda i uzročni) događaj ('odgovor akutne faze') u neuropatologiji AD, a ne samo nespecifičan odgovor na oštećenje neurona. Dva od potencijalno najvažnijih citokina koje luče astrociti, a koji potiču rast DN (a tako i nastanak NP) u AD su S100b protein i bazični fibroblastni činitelj rasta (bFGF, od ‘basic fibroblast growth factor’), što je potvrđeno eksperimentima in vitro i biokemijskom analizom ekstrakata moždanog tkiva sljepoočnog režnja.
Oligodendroglija stanice, kao i astrociti, u AD razvijaju neurofibrilarne promjene, ali se one ultrastrukturalno sastoje isključivo od SF. Demijelinizacija se u AD neuropatološki očituje selektivnim nepotpunim infarkcijama bijele tvari (SIWI, od ‘selective incomplete white matter infarctions’), koje se mogu vidjeti i pomoću MRI. Postepeni nastanak i širenje neurofibrilarnih promjena u AD obrnuto rekapitulira kortikalnu mijelogenezu, nastaje najprije u područjima u kojima se mijelinizacija javila najkasnije (asocijativna područja moždane kore) i postepeno progredira prema područjima gdje se javila najranije (primarna motorička i osjetna područja). Iako nije jasno što bi ovaj nalaz mogao točno značiti, postavljena je hipoteza da oligodendrociti otpuštaju određene, zasad neutvrđene činitelje koji imaju utjecaj na neurone, a poremetnja lučenja ovih činitelja možda dovodi do izrastanja aksonskih mladica (‘sprouting’), poremećaja citoskeleta neurona i nastanka neurofibrilarnih promjena.
2.6.5. Mehanizmi odumiranja i profil vulnerabilnosti živčanih stanica
Otkad su Lucas i Newhouse pokazali da parenteralno davanje glutamata dovodi do degeneracije unutrašnih slojeva retine 1957. godine, a Olney 1969. godine da isti postupak dovodi do masivne degeneracije neurona hipotalamusa i drugih dijelova živčanog sustava postavljajući tako temelje konceptu eksitotoksičnosti, do današnjih je dana potvrđeno nebrojeno puta i na različite načine da je ekscitotoksičnost posredovana glutamatnim (a u manjoj mjeri i AMPA i kainatnim) receptorima, odnosno ekscitatornim aminokiselinama (EAA, od 'excitatory amino acids'), jedan od glavnih patogenetskih mehanizama smrti neurona. Ekscitotoksični proces ima važnu ulogu u poremećajima povezanim s anoksijom, inzultom, hipoglikemijom, epilepsijom i brojnim neurodegenerativnim bolestima, npr. amiotrofičnom lateralnom sklerozom, Huntingtonovom bolešću i naročito AD. Glutamatnim NMDA (N-metil-D-aspartat) receptorima posredovana ekscitotoksičnost rezultat je povećanog utijeka kalcijevih iona uslijed prejake aktivacije NMDA, a u manjoj mjeri i AMPA (od a-amino-3-hidroksi-5-metil-4-izoksazol propionska kiselina) i kainatnih receptora, a kalcijevi ioni u povišenim koncentracijama direktno i indirektno intracelularno pokreću brojne akutne i kronične ekscitotoksične procese koji u konačnici dovode do stanične smrti.
Najvažniji procesi koji posreduju toksičnost glutamata, odnosno EAA, su oni koji su povezani s degeneracijom i promjenama citoskeleta neurona, te oni koji dovode do dugotrajnih promjena morfologije dendritičkih trnova (spina), a najvažniji kandidati koji tvore sponu između povećane koncentracije kalcija i navedenih promjena su proteaze ovisne o kalciju. Dobro je poznato da su u normalnim okolnostima potrebne temeljite strukturalne promjene da bi došlo do rastavljanja i ponovnog sastavljanja elemenata citoskeleta za postizanje određenog oblika spine. Ovaj proces uključuje probavljanje nekoliko vrsta strukturalnih bjelančevina, što se odvija putem kalpaina, tiolnih proteaza koja se aktiviraju kalcijem (imunocitokemijski je dokazano da se nalaze u području postsinaptičkog zgusnuća). Inhibitori kalpaina štite neurone od ekscitotoksičnih i hipoksičnih inzulta, iako ova zaštita nije univerzalna. Postoje mnogobrojni dokazi koji govore u prilog uključenosti kalpain-spektrin interakcija u tijeku dugoročne potencijacije sinapsi (LTP, od ‘long-term potentiation’). Npr. davanje glutamata u ‘kriške’ hipokampusa dovodi do naglog porasta koncentracije spektrina (jedan je od glavnih proteina citoskeleta postsinaptičkog zgusnuća), a infuzija leupeptina (antagonist kalpaina) inhibira indukciju LTP. Također, davanje ekscitotoksičnih količina glutamata ili kainata dovodi do značajnog raspada spektrina u hipokampusu. Iz ovih nalaza proizašla je pretpostavka da je možda aktivacija kalpaina ili sličnih proteaza uzrokom poremećaja citoskeleta, tj. patologije karakteristične za AD. Jedno od takvih spregnutih djelovanja vjerojatno je posredovano fosfoseril/fosfotreonil fosfatazom PP3 (PP2B ili kalcineurin), čija aktivnost zavisi od unutarstanične koncentracije kalcija, a njena prekomjerna aktivacija dovodi do abnormalne fosforilacije tau proteina i nastanka NFT. Druga velika skupina procesa putem kojih povećana unutarstanična koncentracija kalcija dovodi do degeneracije živčanih stanica je prekomjerna aktivacija fosfolipaza (ponajviše fosfolipaze A2) što dovodi do degradacije stanične membrane. U skladu s navedenim, inhibicija PL-A2 djelomično oslabljuje neke oblike ekscitotoksičnog oštećenja. I drugi enzimi koji se aktiviraju kalcijem mogu doprinositi ekscitotoksičnom mehanizmu degeneracije: protein kinaza C, fosfolipaza C, kalcij/kalmodulin ovisna protein kinaza II, neuronalni oblik sintetaze dušičnog monoksida, itd.. Nadalje, visoka unutarstanična koncentracija kalcija dovodi do oštećenja mitohondrijalne membrane, što također ima važnu ulogu u ekscitotoksičnoj degeneraciji. S tim je možda povezana i činjenica da je u bolesnika s AD smanjena aktivnost mitohondrijalnog enzima piruvat-dehidrogenaze. Ovaj enzim sudjeluje u regulaciji homeostaze kalcija u neuronima, pa tako možda nastaje još jedan zatvoreni krug.
Pretpostavki o eksitotoksičnoj degeneraciji neurona u AD govori u prilog činjenica da je glutamat glavni neurotransmiter projekcijskih piramidnih neurona koji čine kortikokortikalne sustave vlakana (kao što je već rečeno, ovi su neuroni selektivno zahvaćeni patološkim procesom u tijeku AD, a stupanj njihovog oštećenja direktno pozitivno korelira sa stupnjem demencije). Također, i drugi piramidni i ne-piramidni neuroni kojima je neurotransmiter glutamat skloniji su stvaranju NFT. U području hipokampalne formacije i entorinalne moždane kore najprije degeneriraju neuroni sloja II entorinalne moždane kore od kojih započinje perforantni put, a njihov je transmiter također glutamat.
U kulturi neurona Ab remeti homeostazu kalcija jer stvara pore u staničnoj membrani koje propuštaju kalcij, čime dolazi do ekcitotoksične smrti. Blokiranje voltažnih kalcijevih kanala s Co2+ ili nimodipinom smanjuje ovaj učinak. Puferiranje koncentracije kalcija putem proteina parvalbumina, kalbindina i kalretinina koje sadrže GABAergički inhibitorni neuroni vjerojatno sprečava njihovu ekscitotoksičnu smrt uslijed disfunkcije glutamatnog sustava. Stoga oni ne odumiru ni u slučajevima AD s visokom učestalošću SP i NFT. U neokorteksu čovjeka ovi neuroni pokazuju visoki stupanj morfološke specifičnosti. Tako su parvalbumin-imunoreaktivni neuroni obično košaraste i stanice u obliku lustera ('chandelier'), kalbindin-imunoreaktivni neuroni su većinom dvostruko-kitičasti ('double-bouquet'), a kalretinin se nalazi u bipolarnim i dvogrmim ('bitufted') stanicama. Dok su parvalbumin i kalretinin imunoreaktivni neuroni naročito otporni i pokazuju dobro očuvanu morfologiju u AD, neki kalbindin-pozitivni neuroni ipak pokazuju određeni stupanj osjetljivosti. Tako je populacija kalbindin pozitivnih interneurona slojeva II i III neokorteksa izuzetno otporna na degeneraciju u tijeku AD, dok su kalbindin pozitivni interneuroni slojeva V i VI nešto manje otporni, pa budu zahvaćeni neurofibrilarnim promjenama u slučajevima s izuzetno velikim brojem NFT. Postoji također manja subpopulacija piramidnih neurona sloja III koji sadrže kalbindin, a koji su u bolesnika s AD također osjetljivi na degeneraciju. U nekim je interneuronima kalbindin kolokaliziran sa somatostatinom (SS). Ciljni neuroni ovih interneurona su vulnerabilni neuroni trećeg sloja, što čini somatostatin i kalbindin imunoreaktivna vlakna, tj. aksonski dio ovih interneurona osjetljivijim na oštećenje u tijeku AD od samih citoplazmi. Shodno navedenom, vlakna imunoreaktivna na somatostatin često su prisutna u SP, dok su sama tijela stanica otpornija na patološke promjene. U tijeku AD, primijećena je također visoka otpornost neurona koji sadrže CRH (hormon koji stimulira otpuštanje kortikotropina), vjerojatno stoga što je u ovim neuronima koji imaju oblik lustera ('chandelier') CRH kolokaliziran s parvalbuminom.
Hipoteza oksidativnog stresa kao uzroka neurodegenerativnih bolesti, temelji se velikim dijelom na poremećajima oksidativnog metabolizma u mitohondrijima, a teoriju starenja organizma uslijed stvaranja slobodnih radikala prvi je predložio Harman 1955. godine. Naime, redukcija kisika u vodu omogućuje stanicama najveći iznos biološki korisne energije. No, vrijednost ovog evolucijskog ‘izuma’ istovremeno ima i jednu manu (koja sa stajališta evolucije nije bitna, jer se događa nakon reprodukcije), a to je da su nepotpuno reducirani kisikovi spojevi (superoksid, peroksidi i drugi) vrlo reaktivni i mogu, ukoliko nisu pod potpunom kontrolom, uzrokovati oštećenja različitih važnih molekula, uključujući i DNA, te u konačnici dovesti do smrti stanice. Navedeni učinak u eukariotičkim stanicama najizraženiji je upravo u mitohondrijima jer se tu odvija najveći dio redukcije kisika. Oštećenja DNA nastala putem djelovanja slobodnih radikala kisika kumuliraju se tijekom života dovodeći do mutacija, disfunkcije i smrti stanica, a možda je upravo zato dob najvažniji rizični činitelj za AD. Ovaj je način oštećenja od naročite važnosti za živčane stanice jer su one jedine, uz mišićne stanice, postmitotičke u trenutku rođenja. Ovakva zakočenost staničnog ciklusa posljedica je činjenice da stečeni obrasci sinaptičkih veza nisu kodirani na razini genoma (i ne mogu se prenositi mitozom), pa bi dioba neurona interferirala s akumulacijom iskustva kroz vrijeme.
Pod oksidativnim stresom podrazumijevaju se posljedice nesrazmjera između produkcije slobodnih radikala i sposobnosti obrane stanice od njihovog djelovanja. Reaktivni radikali kisika (ROS, od ‘reactive oxygen species’, najvažniji su superoksidni anion O2-., hidroksilni radikal OH-., dušični monoksid NO., singularni kisik 1/2 O2., te alkoksilni i peroksilni radikali RO. i ROO., a u širem kontekstu također su bitni i peroksinitritni radikal ONOO-, vodikov peroksid H2O2 i molekularni kisik O2) oštećuju lipide oksidacijom, a proteine i DNA oksidacijom, dekarboksilacijom i deaminacijom, što na kraju završava cijepanjem lanaca DNA. I fragmentacijom Ab nastaju reaktivni peptidni slobodni radikali i superoksidni radikali koji dovode do lipoperoksidacije. ROS mogu biti i na razne druge načine uključeni u patogenezu AD, npr. postoje pretpostavke da oni doprinose nastanku SP potpomaganjem agregacije Ab. Hipotezi oksidativnog stresa govori u prilog i višestruko potvrđen nalaz o oštećene sposobnosti popravka DNA u bolesnika s AD. Veza između nasljednih mutacija dijelova mitohondrijalne DNA (mtDNA) koji kodiraju citokrom oksidaze I i II (CO-I, CO-II) i nastanka AD s kasnim početkom podupire hipotezu poremećaja oksidativnog metabolizma kao mogućeg zajedničkog završnog puta mnogobrojnih molekularnih uzroka AD, budući da su ove mutacije češće u djece majki oboljelih od AD, nego u očeva s AD (što je sukladno naslijeđivanju mtDNA od majke).
Analizom 1092 ispitanika (667 žena i 425 muškaraca, prosječne starosti 76 godina; u razdoblju od 8 godina, 111 osoba je postalo dementno, od toga 83 zbog AD) iz Framinghamske epidemiološke studije, utvrđeno je da povećanje koncentracije homocisteina u plazmi za 5mmol/L povećava rizik za nastanak AD za čak 40%, a povećanje od 14mmol/L povećava rizik za gotovo 200%. Ovaj utjecaj je bio neovisan od starosti, spola, apoE genotipa i drugih poznatih rizičnih činitelja za nastanak AD. Smatra se da je povećana koncentracija homocisteina uzrokom povećanog stupnja oštećenja DNA, jer u eksperimentalnim modelima u kulturama neurona on interferira s popravkom DNA uslijed oksidativnih oštećenja, nastalih između ostalog i djelovanjem Ab. Zbog činjenice da folat smanjuje koncentraciju homocisteina u plazmi, velike se nade polažu u mogućnost da bi njegovo dodavanje u prehranu moglo smanjiti incidenciju AD (od uobičajenih namirnica, folne kiseline ima puno u špinatu), kao što je to već dokazano za arteriosklerozu.
Još uvijek nije jasno da li slabljenje metabolizma glukoze (hipoteza o AD kao ‘dijabetesu mozga’) uzrokuje degeneraciju neurona (koji su, za razliku od drugih stanica, u potpunosti ovisni o njemu) ili pak degeneracija neurona uslijed drugih uzroka dovodi do remećenja metabolizma glukoze. Bilo kako bilo, to je jedna od najranijih promjena koja se može ustanoviti u bolesnika s AD. Razina molekula transportera glukoze smanjena je u osoba s AD, a naročito redukcija GLUT3 transportera bila je veća od stupnja gubitka sinapsi, što ukazuje da je ovo smanjenje mogući uzrok, a ne posljedica oštećenja metabolizma glukoze u AD. U neurona koji imaju problema s energijskim metabolizmom i normalne količine glutamata mogu dovesti do ekscitotoksičnog oštećenja. Još jedan važan element slabljenja energijskog metabolizma u AD je stanje kapilara, jer transportni sustavi u njihovim zidovima mogu biti oštećeni uslijed deponiranja amiloida.
Na temelju pretežno morfoloških kriterija može se razlikovati nekoliko vrsti
smrti stanica: apoptoza (tip 1), autofagička degeneracija (tip 2), ne-lizoosmalna
disintegracija (tip 3A) i ne-lizosomalna citoplazmatska degeneracija (tip 3B).
Tipovi 3A i 3B se zajedničkim imenom nazivaju nekrotičnom smrcu. Programirana
smrt stanice (PCD, od 'programmed cell death') je važna, genetski visoko konzervirana
biološka strategija za uklanjanje nepoželjnih stanica organizma (viška stanica
stvorenog tijekom razvitka, oštećenih ili inficiranih stanica), a apoptoza jedan
od njenih najvažnijih modusa. Patofiziološki, apoptoza je aktivni, brz i obično
svrsishodan oblik stanične smrti ('samoubojstvo' stanice), dok je nekroza nesvrsishodan,
uglavnom pasivan i puno dugotrajniji proces koji nastupa uslijed različitih
oblika oštećenja stanice. Apoptoza najčešce zahvaća samo pojedinačne stanice,
a nekroza skupine oštećenih stanica. Morfološki, stanice koje umiru apopotičkom
smrcu imaju brojne protruzije svoje površine, a uslijed kondenziranosti citoplazme
i jezgre (piknoza) se skvrčavaju, dok nekrotične karakterizira ruptura i otapanje
većine organela i membrana, uključujući i citoplazmatsku. U tijeku apoptoze
kromatin bude pocijepan putem aktiviranih endogenih endonukleaza (npr. DFF40,
od DNA fragmentation factor 40 - DFF40, naziva se još i CAD, od 'caspase-3 activated
DNAse', zatim DNAza II, DNAza g, NUC endonukleaze različite molekularne težine,
DNAS1L3 i druge) u fragmente oligonukleosomalne duljine (u zavisnosti od vrste
stanica i eksperimentalnog modela izmedu 50 i 300 kilo parova baza, najčešće
oko 180-200 kbp ili višekratno toliko; fragmenti su te duljine zato jer endonukleaze
cijepaju eksponirane dijelove DNA izmedu histona) što se elektroforezom lijepo
prikazuje kao niz ravnomjerno rasporedenih pruga koje stoga sliče ljestvama
('DNA ladder'). Za razliku od toga u nekrozi se na elektroforetskom gelu vidi
samo 'šmir', a ne pruge, što ukazuje na slučajno cijepanje DNA u fragmente različite
duljine. Naposlijetku, u apoptozi nastupa odjeljivanje (kompartmentalizacija)
i jezgrinih i citoplazmatskih dijelova u 'apoptotička tijela' različitog oblika
i veličine, a mnoga od apoptotičkih tijela sadrže fragmente jezgre (tj. kromatina).
Iako je cijela stanica skvrčena, cisterne endoplazmatkog retikuluma su raširene.
Stoga se čini da se putem endoplazmatskog retikuluma odvodi voda, što daje stanici
vakuolizirani izgled površine.
Endonukleaze odgovorne za cijepanje DNA tijekom apoptoze aktiviraju se uslijed
razgradnje svojih inhibitora (npr. DFF45 ili ICAD, od 'inhibitor of caspase-3
activated DNAse'). Glavnu skupinu molekula koje razgrađuju inhibitore endonukleaza
čine kaspaze, golema obitelj cisteinskih proteaza. Putevi aktivacije kaspaza
nisu još ni izdaleka precizno upoznati, no zasad se može reći da postoje proteini
koji promoviraju aktivaciju kaspaza (npr. bclxs, bax, bad, bak, bik i drugi
proapoptotički članovi obitelji bcl-2 proteina), te oni koji je inhibiraju (npr.
bcl-2, bclxl, bhrfl i drugi antiapoptotički članovi bcl-2 obitelji proteina,
te obitelj proteina inhibitora apoptoze (IAP) npr. XIAP, ML-IAP-1, cIAP-1 i
2, NAIP, survivin, itd.). Glavna razlika izmedu bcl-2 i IAP obitelji proteina
je ta što bcl-2 obitelj proteina djeluje na mitohondrijskoj membrani regulirajući
otpuštanje citokroma c, dok IAP proteini većinom direktno inhibiraju kaspaze
koje se nalaze u citoplazmi. Kao odgovor stanice na npr. gubitak trofičke potpore,
dolazi do prevage ekspresije proapoptotičkih bcl-2 proteina u mitohondrijskoj
membrani i otpuštanja citokroma c iz mitohondrija (nije još jasno da li se to
dogada uslijed promjene permeabilnosti mitohondrijske membrane, putem polaritet
ovisnih anionskih kanala mitohondrija ili pak aktivacije ionskih kanala u vanjskoj
mitohondrijalnoj membrani), što potiče stvaranje APAF-1 ('apoptotic protease
activating factor 1') apoptosoma koji zatim točno određenim slijedom aktivira
različite kaspaze, od kojih se među najvažnije 'izvršne' ubrajaju kaspaze 9,
3 i 7. Da apoptotički proces može napredovati i bez vidljive internukleosomalne
fragmentacije DNA dokazuju pokusi u kojima izolirane jezgre eksponirane estraktima
citosola apoptotičnih stanica pokazuju kondenzaciju i degradaciju kromatina
kao u apoptozi, te činjenica da i enukleirane stanice mogu pokazivati morfološke
promjene karakteristicne za apoptozu. Za razliku od nekrotične smrti gdje je
disfunkcija mitohondrija karakterističan poremećaj, mitohondriji u apoptozi
obično ne pokazuju ranih promjena i ostanu do kraja funkcionalno aktivni (potrebna
je energija za izvršenje samoubojstva gotovo do samog kraja). Također, za vrijeme
apoptoze karakteristično ne dolazi do rasipanja DNA, lizosomalnih enzima, oksidiranih
lipida i drugih katabolita u međustanični prostor, te nisu aktivirani upalni
mehanizmi, kao što je to slučaj pri nekrotičnoj smrti stanice. Posljednje ostatke
apoptotički umrlih stanica odstranjuju fagocitozom bilo susjedne stanice, bilo
solitarni rezidentni makrofagi, tj. mikroglija stanice.
Gubitak neurona je vrlo izraženo obilježje AD, naročito u entorinalnoj moždanoj
kori i hipokampalnoj formaciji. Nekoliko je grupa istraživača predložilo, većinom
na temelju in vitro eksperimenata, označavanja fragmentiranih dijelova DNA i
imunocitokemijskih postupaka za dokazivanje proapoptotičkih proteina, da u tijeku
AD neuroni mogu degenerirati apoptotičkim procesom. No, osim u podrucju GVD
CA1 stanica (tzv. lokalna apoptoza), u tijeku AD vidi se isključivo nekrotična
smrt. Ovaj stav se temelji na činjenici da je pozitivni nalaz gore navedenih
metoda vjerojatno posljedica postmortalnih procesa, da se ne vidi tipična apoptotička
morfologija neurona, a takoder elektroforezom nije moguće dobiti karakteristične
fragmentacijske pruge. Također, ako se uzme u obzir kinetika apoptotičke smrti,
tj. da se sam proces traje 2-7 sati, te da se apoptotička morfologija može vidjeti
samo kroz 3 do najviše 72 sata, kad bi doista veliki broj pozitivnih stanica
koje se vide na tankim rezovima moždanog tkiva degenerirao apoptozom, to bi
značilo da bi cijela promatrana struktura degenerirala u roku nekoliko dana,
što je malo vjerojatno u jednoj dugotrajnoj bolesti kao što je AD. No, budući
da u tijeku AD doista dolazi do porasta ekspresije nekih proapoptotičkih mRNA,
to ukazuje na cinjenicu da neke stanice ipak pokušavaju izvršiti 'samoubojstvo'
(ali u tome ne uspijevaju). Ovakav je nalaz nazvan abortivnom apoptozom (ili
'abortozom'). Na tragu ovog nalaza je i još jedna u nizu novijih fascinirajućih
hipoteza, a to je da je u patogenezi AD važan porast gonadotropina ulijed menopauze/andropauze,
što pokreće mehanizme izlaska iz Go faze mirovanja staničnog ciklusa, a ova
dediferencijacija i reaktivacija morforegulatornih gena možda dovodi do tau
i amiloidne patologije (npr. neuron koji se hoće podijeliti mora najprije hiperfosforilirati
tau proteine da bi razdvojio postojeće mikrotubule). U prilog ovoj pretpostavci
govore činjenice da naročito puno receptora za LH imaju baš hipokampalni neuroni,
te da, u usporedbi s kontrolama, bolesnici s AD imaju povećane koncentracije
FSH i LH. Takoder, sve je više podataka o povećanoj aktivaciji drugih mitogena
i njihovih receptora u ranim stadijima AD. U liječenju AD potencijalno bi se
vrlo skoro mogao stoga pokušati koristiti leuprolid koji snižava razinu LH,
a već se koristi u liječenju karcinoma prostate.
Za razliku od NFT i SP, gubitak neurona odraz je kumuliranog oštećenja tkiva
(kao rezultat i starenja i AD). Zato pri interpretaciji kvantitativnih podataka
koji se odnose na gubitak živčanih stanica treba imati na umu da je smrt neurona
samo zajednički završni put sveukupnih patoloških procesa (i AD i starenja),
a promatrana patološka obilježja većinom relativno kasne posljedice ovih procesa.
Takoder, treba znati da odnos izmedu količine i raspodjele NFT, gubitka neurona
i demencije nije linearan. Ovo se djelomično može pripisati činjenici da se
zbog redundancije živčanih elemenata, te kompenzatornih strukturalnih, fizioloških
i biokemijskih procesa do određene granice gubitak neurona tolerira bez funkcionalnih
posljedica. Tako je npr. opisan rast dendritičkog stabla u poodmakloj dobi,
vjerojatno kao kompenzacija za gubitak neurona, te kolinergička reinervacija
deaferentiranih područja završetka perforantnih vlakana. Također je poznato
da neuroni s NFT kompenzatorno imaju veće dendriticko razgranjenje (s još uvijek
aktivnim sinapsama) od susjednih neurona bez NFT.
2.7. Dosadašnji pokušaji liječenja i trenutni klinički pokusi
U svakodnevnoj kliničkoj praksi, liječenje AD je simptomatsko. Do sada su u kliničkim pokusima najviše korišteni i najbolje proučeni kolinomimetici, najčešće inhibitori acetil-kolinesteraze (AChE), npr. takrin, donepezil, galantamin, fizostigmin, eptastigmin, rivastigmin, velnakrin, metrifonat, MSF (metansulfonil fluorid), ksanomelin, sabkomelin i drugi, ali također u manjoj mjeri i kolinergički agonisti arekolin, RS-86 (2-etil-8-metil-2,8-diazospiro-1,3-dianhidrobromid), AF serija analoga acetilkolina (najčešće AF102B), oksotremorin, pilokarpin, ksanomelin, cevimelin, milamelin, sabkomelin, besipirdin, cinopirdin, betanehol (budući da ne prolazi krvno-moždanu barijeru davan je intraventrikularno), nikotin i nikotinski agonist ABT-418. Općenito, takrin (tetrahidroaminoakridin) i ostali inhibitori kolinesteraze, kao i kolinergički agonisti, samo prolazno i kratkotrajno usporavaju progresiju demencije i to samo u nekih bolesnika u ranom stadiju bolesti. Temeljeći njihovu uporabu na nalazu deficita kolinergičke transmisije u AD, eksperimentiralo se i sa davanjem prekursora acetilkolina: kolina i lecitina (odnosno njihovim dodavanjem u prehranu). Ni ovi rezultati nisu dali ohrabrujuće rezultate: kolin nije imao baš nikakvog utjecaja na kognitivne funkcije, dok je lecitin imao samo vrlo blage i u manjeg broja pacijenata. Iz istog su razloga prekinuta klinička ispitivanja idebenona (antioksidans), suritozola (inverzni GABA agonist), propentofilina (blokira ponovni unoz adenozina i inhibira fosfodiesterazu) i acetil-1-karnitina (pretpostavljalo se da će povećati djelotvornost mitohondrija).
Gotovo 90% svih kolinergičkih neurona bazalne jezgre mozga čovjeka pokazuje imunoreaktivnost za beta podjedinicu živčanog činitelja rasta (p75-NGFr, ‘low affinity nerve growth factor receptor’). Iako p75-NGFr ima vrlo važnu ulogu u aktivnosti i djelovanju neurotrofina, ovaj signalni mehanizam također ovisi i o istovremenoj interakciji s visokoafinitetnim trk (‘tyrosine kinase high-affinity receptors’) receptorima. p75-NGFr i trk receptori (trk-A, trk-B i trk-C) se anterogradno transportiraju do moždane kore gdje se na njih veže NGF, ali također i drugi neurotrofini iz NGF 'superobitelji'. Kompleks NGFr-NGF/trk se zatim retrogradno transportira do kolinergičkih neurona bazalne jezgre, čija je ChAT i AChE aktivnost izravno ovisna o NGF. Stoga uslijed dugotrajnog gubitka NGF dolazi do njihovog potpunog odumiranja. Pretpostavlja se da NFT i SP interferiraju s prijenosom NGF, odnosno njegovim vezanjem za NGFr/trk i retrogradnim transportom u bazalnu jezgru, pa bi zato rana selektivna vulnerabilnost bazalne jezgre u AD mogla biti posljedica činjenice da je bazalna jezgra jedina subkortikalna jezgra u mozgu odraslog čovjeka koja pokazuje imunoreaktivnost za NGFr. Pod ovom pretpostavkom je najprije u jednoga, a zatim u još dvoje bolesnika oboljelih od AD u terapijske svrhe intraventrikularno apliciran NGF. No, i pored nekih ohrabrujućih povoljnih učinaka na kognitivni status ovih bolesnika, zbog jakih nuspojava NGF-a, odustalo se od njegove daljnje primjene.
Tri godine nakon što su 1999. godine Schenk i suradnici iz farmaceutske tvrtke Elan objavili da je aktivna imunizacija pomoću Ab u transgeničnih miševa koji prekomjerno eksprimiraju mutirani (valin na poziciji 717 zamijenjen fenilalaninom) AbPP čovjeka dovela do smanjivanja stvaranja SP, DN i astroglioze nakon 6 tjedana odnosno 11 mjeseci po imunizaciji, farmaceutske tvrtke Elan i American Home Products ishitreno su i nepromišljeno pokušale na identičan način imunizirati i ljude oboljele od AD (pasivna imunizacija anti-Ab protutijelima nije pogodna jer bi se morala vršiti svaka 3 tjedna zbog kratkog poluživota IgG protutijela, što je neizvedivo kod kronične bolesti kakva je AD, a to bi dovelo i do anafilaktičke reakcije odnosno serumske bolesti; eventualno bi ovakav pristup možda bio moguć pomoću samo jednog lanca protutijela što sadrži mjesto za prepoznavanje antigena). Najprije je sintetskim Ab (vakcina AN-1792) imunizirano 80 bolesnika u srpnju 2001. godine, a zatim još 280 ljudi u Sjedinjenim Američkim Državama, Švicarskoj, Španjolskoj, Velikoj Britaniji i Francuskoj. Budući Ab prolazi krvno-moždanu barijeru, glavna ideja je bila da se protutijelima ‘izvuče’ suvišni Ab (tzv. ‘peripheral sink hypothesis’), kako se ne bi odlagao u moždanom tkivu. Nažalost, najprije su 11. siječnja 2002. godine četiri bolesnika (od 79) u Francuskoj, a ubrzo zatim još i 15 bolesnika u drugim državama razvili simptome encefalitisa ili meningoencefalitisa, nakon čega je 23. siječnja 2002. godine imunizacija prestala, a studija službeno okončana 1. ožujka 2002. godine. Budući da nije pronađena korelacija između titra anti-Ab protijela i nastanka meningoencefalitisa, pretpostavlja se da je upalni process nastao posredovanjem limfocita T. Iako je Elan tvrdio suprotno (a za ‘nuspojavu’ okrivio reaktivaciju herpes simpleks tip 1 virusa u navedenih bolesnika), za većinu istraživača ovo je bio očekivani događaj, jer je u svrhu imunizacije upotrijebljena cijela Ab1-42 molekula. Naime, i prije provedbe aktivne imunizacije bilo je dobro poznato da upalnu reakciju moždanog tkiva mogu izazvati i Ab i protutijela koja se na nj stvaraju, da dulji oblik Ab in vitro agregira za samo 20 minuta, da je najskloniji fibrilogenezi, te da je za antigeničnost je bitan samo amino-terminalni kraj Ab1-16 koji ne izaziva upalnu reakciju. Dulji oblik također reducira specifičnost stvorenih protutijela. Povrh toga, doslovce na desetke istraživanja ukazuje na zaštitna svojstva Ab, uključujući ona anti-oksidativna (npr. pri niskom pH, Ab veže ione bakra i zinka, a kad acidoza nestane ih otpušta – što objašnjava zašto se u akutnoj fazi traume mozga kao fiziološki odgovor stvara puno Ab ili npr. kad se doda rekombinantni AbPP u medij neurona Down sindroma, i do 3 puta više stanica preživi različite oblike staničnog stresa), te kao molekule koja smanjuje propusnost krvno-moždane barijere (nakon njegovog odstranjivanja protutijelima logično je bilo očekivati da će doći do ‘curenja’ pojedinih komponenti plazme u moždano tkivo). Dakle, AN-1792 je napravljen da izazove specifičan autoimuni odgovor na deponirajući Ab, no pritom se nije mislilo na notornu činjenicu da se on ne nalazi samo u SP, već i u izvanstaničnoj tekućini. Kao što je već navedeno, ubikvitarnost ovog peptida u normalnih osoba ukazuje na činjenicu da on nije neurotoksičan u svom topljivom obliku, a istraživači koji podržavaju hipotezu o neurotoksičnosti Ab nikad nisu objasnili zašto bi se, protivno temeljnim biološkim zakonitostima, uopće proizvodio takav jedan ‘toksin’ u tolikoj količini: glutamat i amonijak u povećanoj količini vrlo su toksični, ali nikom nije palo na pamet odstranjivati ih iz mozga jer bi to zasigurno dovelo do poremećene funkcije neurona uslijed njihove povezanosti u intermedijarnom metabolizmu. Kolektivna opsesija i surova borba za prevlast na farmaceutskom tržištu dovela je do toga da se previdi ono očigledno: ovaj peptid sigurno se ne bi proizvodio u tolikoj količini da njegova toksičnost nije manja od koristi za djelovanje mozga. Osim toga, čak i kad bi se riješile ‘nuspojave’ izazivanja prejake upalne reakcije mozga na imunizaciju, te smanjila propusnost krvno-moždane barijere, nije riješen problem eliminacije topljivog Ab, budući da nisu poznate njegove normalne funkcije. Greška je također učinjena jer se vakcinacija ljudi temeljila samo na nalazima u transgeničnih miševa s prekomjernom ekspresijom APP čovjeka. Protutijela su u ovih miševa očigledno ‘očistila’ humani AbPP, ali je koncentracija endogenog Ab miševa ostala nepromijenjena. Trebalo je najprije napraviti odgovarajuće kontrolne eksperimente, koji su trebali uključivati normalne miševe ili još bolje primate i njihov endogeni Ab, što nije učinjeno.
Trenutno su u različitim fazama farmakoloških i kliničkih ispitivanja inhibitori AChE (huperzin A, početna faza, Kina; fenilkarbamat fizostigmina fenserin, II faza, Axonyx); kolinergički agonisti (nefiracetam, II faza, Daichii; ksanomelin, II faza, Eli-Lilly); estrogeni (faza III), antioksidansi, npr. alfa-tokoferol (vitamin E), i L-deprenil (inhibitor monoaminooksidaze B); nesteroidni antireumatici, npr. ibuprofen (faza III, American Home Products, Upjohn, Bristol-Myers Squibb) i naproksen (faza III, Syntex, Proctor&Gamble, Roche); statini npr. atorvastatin, lovastatin, pravastatin, cerivastatin, fluvastatin, simvastatin: jedno veliko istraživanje na 60000 bolesnika koji su koristili atorvastatin (konvertira 3-hidroksi-3-metil-glutaril-koenzim A u mevalonat, čime sprečava biosintezu kolesterola) pokazalo je statistički značajno manju prevalenciju AD u tih osoba, a ovi lijekovi također blokiraju aktivaciju limfocita T putem interferona g; AIT-082 u cerebrolizinu (III faza, Ebewe), odnosno neotrofinu (III faza, Neotherapeutics) što aktivira gvanilil-ciklazu koja potiče proizvodnju neurotrofina i aktivnost hem-oksigenaze; protuupalni lijekovi, npr. rofekoksib (inhibitor ciklo-oksigenaze 2, faza II, Merck), valproat (stimulator GABA receptora, faza III); dapson (inhibira djelovanje neutrofila, faza II, Immune Network); melatonin (kliničke studije u AD tek su u pripremi; u žutom tisku se reklamira kao ‘čudotvorni lijek’ za sve bolesti i starenje, no za to nema znanstvenih potvrda; u Sjedinjenim Američkim Državama se smatra dodatkom prehrani pa se može kupiti i bez recepta, dok je u Europskoj Uniji klasificiran kao neurohormon i ne može se kupiti); ampakin (1-(kvinoksalin-6-ilkarbonil)piperidin (modulira djelovanje AMPA receptora, Cortex, III faza); memantin (antagonist NMDA receptora, III faza, Merz); ekstrakti različitih biljaka (npr. EGb 761 antioksidativni ekstrakt ginkgo bilobe, te japanske biljke utan-kampo koja izgleda poiče proizvodnju neurotrofnih činitelja), te inhibitori beta i gama sekretaze (početna faza, Amgen, Bristol-Meyer Squibbs, Elan, Scios, SmithKline Beecham). Također se planira i kontrolirano dodavanje folata u prehranu (jer smanjuje koncentraciju homocisteina u plazmi).
3. Ostali češći uzroci demencije
Pod dijagnozom vaskularne demencije (VaD) podrazumijevaju se bolesnici u kojih se različiti cerebrovaskularni incidenti ili patološke promjene krvnih žila mozga očituju kao dominantan uzrok kliničke slike demencije. U vrijeme kad je sifilis bio vodeći uzrok demencije, aterosklerotsku demenciju opisali su Binswanger 1894. i Alzheimer 1895. godine, a lakunarnu demenciju (‘état lacunaire’) Marie 1901. godine. Kroz veći dio XX stoljeća mislilo se da demencija nastaje isljučivo uslijed smanjene perfuzije mozga. Kad je AD prepoznata kao vodeći uzrok demencije sredinom 70-tih, znanstveni je interes za demenciju vaskularnog porijekla splasnuo. Pa ipak, upravo u to vrijeme Hachinski predlaže naziv demencija s višestrukim žarištima (MID, od ‘multi-infarct dementia’) kako bi što vjernije opisao demenciju koja nastaje uslijed višestrukih tromboembolijskih infarkata (promjera većeg od 15mm; kod lakunarne demencije infarkti su veličine 2-15mm, a oni manji od 2mm nazivaju se mikroskopskim infarktima), naglasio da su broj i smještaj infarkata važniji od volumena oštećenog moždanog tkiva, te predložio ljestvicu za njeno dijagnosticiranje (tzv. HIS ljestvica, od ‘Hachinski ischemic score’). HIS je zapravo načinjen u pokušaju da se olakša dijagnostika VaD u odnosu na AD, a ne sadrži kriterije za dijagnozu demencije per se. Iako je dobro što je HIS utemeljen na konceptu MID jer je to najčešći oblik VaD, ljestvica nije dobra u otkrivanju drugih podvrsti VaD. U HIS se različitim obilježjima pridodaju bodovi, i to po 2 boda za: nagli početak simptoma, fluktuirajući tijek, pozitivnu anamnezu na moždani udar, žarišne neurološke simptome i žarišne neurološke znakove, te po 1 bod za: polaganu progresiju, smetenost u mraku, relativnu sačuvanost osobnosti, depresiju, somatske simptome, emocionalnu inkontinenciju, hipertenziju i aterosklerozu. Ukupni zbroj veći ili jednak 7 govori u prilog VaD, 5 ili 6 miješanoj demenciji (VaD + AD), a manji ili jednak 4 u prilog AD.
U novije vrijeme je koncept VaD značajno proširen, te uključuje višestruke patofiziološke mehanizme povezane s deficijencijom moždanog tkiva u opskrbi krvlju i različitim vrstama patologije kao što su: višestruki infarkti koji nastaju uslijed ateroskleroze velikih krvnih žila i embolije, solitarni strateški infarkti, lakunarni infarkti i promjene bijele tvari uslijed bolesti malih krvnih žila (SVD, od ‘small vessel disease’), hipoperfuzija, intermitentna ishemija, abnormalna vaskularna permeabilnost, hemoragija, hipertenzivna vaskulopatija, ishemija nakon rupture aneurizme, arteritis uslijed različitih autoimunih i infektivnih poremećaja, kolagenoze, itd.. Donešeni su i mnogobrojni novi kriteriji za dijagnozu VaD, od kojih su najpoznatiji kriteriji po ADDTC (od ‘Alzheimer’s Disease Diagnostic and Treatment Centers’) donešen 1992. godine, NINDS-AIREN (od ‘National Institute for Neurological Disorders and Stroke - Association Internationale pour la Recherche et l’Enseignement en Neurosciences’) kriterij donešen 1993. godine, te ICD-10 i DSM-IV kriteriji iz 1994. godine. Relativno slabi rezultati ovih poboljšanih kriterija posljedica su činjenice da je VaD ‘previsoki’ zajednički nazivnik za mnogobrojne procese što dovode do različitih patoloških oblika bolesti, pa se i ne može očekivati da će isti klinički kriterij imati isti uspjeh za sve podvrste AD. Ne čudi zato što su ovi dijagnostički kriteriji za vaskularnu demenciju (VaD) istovremeno prerestriktivni (jer npr. zahtijevaju i prisutnost osjetnih ili motoričkih deficita) i prepermisivni (jer npr. uključuju i bolesnike s kognitivnim deficitima uzrokovanim jednim jedinim ‘strateški smještenim’ infarktom).
Budući da klinička slika bolesti uvijek odražava anatomsku raspodjelu oštećenja, VaD može sličiti drugim primarnim demencijama. Poremećaji funkcije čeonog režnja vjerojatno su najčešća klinička slika VaD iako sam prefrontalni korteks nije zahvaćen patološkim promjenama. Općenito se može reći da izolirana tipična VaD (tj. MID), za razliku od AD, ima rane simptome poremećaja motorike i percepcije, javlja se u mlađoj dobi (između 40 i 50 godina starosti, a onda opet češće poslije 70 godina starosti), pamćenje je slabije oštećeno, sveukupno češće obolijevaju muškarci, obično nastaje naglo, ima tendenciju epizodičke progresije i fluktuacija u tijeku bolesti koja može trajati i do desetak godina, često je povezana sa povećanim krvnim tlakom, koronarnom ili kardiovaskularnom bolešću, žarišni neurološki znakovi se javljaju rano u tijeku bolesti, a trombotički, embolički ili hemoragički infarkti javljaju se pretežno u sivoj tvari.
Dijagnoza VaD postaje malo vjerojatna ako MRI ne pokaže oštećenja koja odgovaraju kliničkoj slici. CT i MRI s velikom sigurnošću prikazuju žarišta hiperintenziteta i infarkte koji su najčešće smješteni u bazalnim ganglijima (putamen i palidus u oko 61% slučajeva, kaudatus 41%), talamusu (27%), ponsu (25%), kapsuli internoj (8%), cerebelumu (8%), korpusu kalozumu (3%), kao i hiperintenzitete bijele tvari čeonog režnja (oko 22%). PET otkriva višestruka žarišta hipometabolizma (za razliku od rane AD gdje je karakteristično smanjen metabolizam u sljepoočnih režnjeva). Funkcijske pretrage u bolesnika s VaD obično pokazuju difuzno i asimetrično smanjenje protoka krvi, a gubitak cerebralnog vazomotornog odgovora redovit je nalaz (za razliku od AD gdje je cerebralni protok krvi reduciran u temporalnim i parijetalnim područjima, ali bolesnik ima sačuvanu sposobnost vazodilatacije i povećanja cerebralnog protoka krvi pri odgovoru na različite podražaje).
Višestruki kortikalni i subkortikalni okluzivni infarkti povezani su s hipertenzijom, srčanom bolešću, arteriosklerozom i dijabetesom. Bolesti miokarda, hipoglikemija, i stanja hipoperfuzije mogu dovesti do kliničke slike demencije uslijed degeneracije CA1 neurona hipokampusa, čak i ako nema dokumentiranih epizoda aresta ili hipotenzije. Binswangerova bolest također može uzrokovati VaD. Glavne patološke promjene u ovoj bolesti su hijalinizacija i zadebljavanje arteriola, što uzrokuje ishemička i sekundarna demijelinizacijska oštećenja bijele tvari. Rizični činitelji uključuju hipertenziju, policitemiju, hiperlipidemiju i hiperviskozitet. Slični rizični činitelji vrijede i za izolirana oštećenja periventrikularne bijele tvari (leukoaraioza). Ove promjene mogu utjecati na kognitivne funkcije, ali su rijetko same po sebi primarnim uzrokom demencije. Mutacija NOTCH-3 gena na kromosomu 19 dovodi do disfunkcije epidermalnog činitelja rasta, abnormalnosti glatkih mišićnih stanica arteriola i nakupljanja PAS-pozitivne tvari u njihovoj stijenci, što dovodi do cerebralne autosomno-dominantne arteriopatije sa subkortikalnim infarktima i leukoencefalopatijom (CADASIL, od ‘cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy’). Ovo je rijedak, ali vjerojatno ‘najčišći’ oblik VaD. Često je povezan s migrenom, a u nekim slučajevima klinička slika može biti vrlo slična AD. Uslijed izazvanih infarkcija i krvarenja i amiloidna angiopatija može uzrokovati VaD (što doprinosi čestoj istovremenoj prisutnosti VaD i AD).
Kad vaskularne lezije zahvaćaju pretežno subkortikalnu bijelu tvar nastaje karakteristični subkortikalni sindrom, subkortikalna arteriosklerotska demencija, koju treba razlikovati od subkortikalnih neurodegenerativnih bolesti i komunicirajućeg hidrocefalusa (budući je proširenje ventrikula uobičajeno obilježje bolesti – tzv. senilni hidrocefalus). Ovaj je poremećaj najčešće povezan s pušenjem i dugotrajnom hipertenzijom. Povećanje ventrikulskog prostora dovodi i do tzv. kribriformnih oštećenja, što su još 1901-1902. godine opisali Marie i Ferrand (‘état criblé’), a radi se o proširenju perivaskularnih prostora.
Proučavanja blizanaca pokazuju da su za nastanak VaD (a za razliku od AD), način života i okolišni činitelji važniji uzročni činitelji od naslijeđa. Stoga se i liječenje VaD treba temeljiti na prevenciji: liječenju hipertenzije (smanjuje incidenciju VaD za 36-50%), prestanku pušenja, kontroli dijabetesa, davanju antikoagulansa kod atrijske fibrilacije, karotidnoj endarterektomiji u simptomatičnih bolesnika sa stenozom većom od 70%, davanjem aspirina pacijentima s visokim vaskularnim rizikom.
Kad je VaD već počela, cilj je liječenja usporavanje njenog napredovanja, pospješivanje kognitivnih funkcija i prevencija nastanka novih infarkata. Dobrim su se pokazali davanje 325mg aspirina dnevno, nimodipin i nikardipin (antagonisti kalcijevih kanala s dilatatornim učinkom), nicergolin (derivat alkaloida žiška s trombolitičkim i dilatatornim djelovanjem) i dodavanje vitamina E u prehranu. Još uvijek je upitna vrijednost propentofilina (inhibitor fosfodiesteraze i ponovnog unosa adenozina, faza III) i memantina (antagonist NMDA receptora, faza III), a pentoksifilin (predmnijevalo se da povećava kapilarni protok), denbufilin (inhibitor fosfodiesteraze) i EGb 761 ekstrakt ginkgo bilobe nisu u dvostruko slijepim kliničkim pokusima pokazali značajne pozitivne učinke.
3.2. Frontotemporalna demencija
Žarišne (lobarne) atrofije obuhvaćaju skupinu primarnih demencija kojima je glavno obilježje ograničeno područje atrofije moždane kore. Ovo područje može biti maleno reda veličine pojedinačnog girusa, a može biti i toliko veliko da zahvaća oba čeona i sljepoočna režnja. Kliničku sliku i neuropatološke promjene frontotemporalne demencije (FTD, od ‘frontotemporal dementia’) prvi je detaljno opisao Pick 1892. godine. U zavisnosti od anatomske raspodjele patoloških promjena, a pod širim pojmom FTD može se razlikovati više kliničkih sindroma. Prilikom simetrične zahvaćenosti obaju čeonih i prednjih dijelova sljepoočnih režnjeva nastaje tzv. tipična FTD, asimetrično zahvaćanje samo dominantne hemisfere dovodi do sindroma primarne progresivne afazije (PPA, od ‘primary progressive aphasia’), asimetrično zahvaćanje desne hemisfere dovodi do psihotičnih simptoma, nekontrolirane impulzivnosti i disinhibicije, pretežno zahvaćanje obaju temporalnih režnjeva dovodi do sindroma tečne (‘stražnje’) afazije s asocijativnom vidnom agnozijom (budući da bolesnik ne može shvatiti verbalne, a zatim ni vidne sadržaje, ovaj se poremećaj naziva i semantička demencija), a amiotrofija može biti povezana sa svim navedenim oblicima FTD.
Sve do sredine 90-tih godina XX stoljeća, neuropatološka definicija FTD temeljila se na prisutnosti jasno ograničene atrofije čeonog i prednjeg dijela sljepoočnog režnja i nespecifičnih histopatoloških promjena. Napredak u genetici, biokemiji i molekularnoj-biologiji u posljednjih je nekoliko godina doveo do radikalnih promjena omogućivši jasno prepoznavanje najmanje četiriju bioloških podvrsti frontotemporalnih demencija: tipičnu FTD, dvije čiste tauopatije (FTDP-17 i Pickovu bolest), te frontotemporalna demencija s bolešću motoneurona. Ipak, sve dok neuropsihološkim, neuroimaging i biokemijskim pokazateljima ne postane moguće donijeti i pouzdanu premortalnu dijagnozu ovih podvrsti FTD, preporučuje se upotreba šireg pojma FTD.
U tipičnoj FTD neuropatološka obilježja su umjerena do jaka atrofija moždane kore ograničena na čeona i temporoparijetalna područja, te proširenje ventrikula. Atrofija je simetrična, no opisani su i asimetrični slučajevi. U područjima atrofije izražen je gubitak piramidnih neurona slojeva II i III, dok su oni u sloju V slabije zahvaćeni. Spongioformna degeneracija slojeva II i III često se vidi zajedno s umjerenom do jakom astrocitnom gliozom u sivoj i bijeloj tvari, te izblijedjelim mijelinom bijele tvari atrofičnog dijela korteksa. Ove se promjene mogu proširiti i na inzulu i girus cinguli. Katkad su prisutni ‘balonirani’ neuroni koji u području ‘balona’ sadrže nakupljene fosforilirane neurofilamente. Apikalni dendriti piramidnih neurona sloja III mogu biti abnormalno tortuotični i bez spina. Zanimljivo je da je kolinergička inervacija sačuvana, dok su somatostatinski i kalbindinski interneuroni uglavnom odumrli. Omjer učestalosti između AD i tipične FTD kreće se između 1:4 i 1:10. Početak FTD javlja se gotovo uvijek prije 65. godine starosti (obično između 45 i 65 godina starosti), a podjednako zahvaća oba spola. Trajanje bolesti je često protrahirano i uobičajeno iznosi 10-15 godina. Usprkos razlikama u topografiji patoloških promjena u pojedinih bolesnika, ključni kriterij za dijagnozu tipične FTD je odsutnost tau- i ubikvitin-pozitivnih unutarstaničnih nakupina, kao što su NFT u AD (ali ne ograničeni broj NFT uobičajen za dob), argirofilna zrnca, Pickova ili Lewyjeva tjelešca. Tipična FTD nema nakupina u gliji, u elektroforetskom profilu vidi se svih 6 izo-oblika tau proteina s prugama od 55, 64 i 69 kDa.
Primarnu progresivnu afaziju (PPA, od ‘primary progressive aphasia’) prvi je opisao Mesulam 1982. godine. PPA je karakterizirana postepenim i relativno izoliranim propadanjem jezičnih funkcija, a dijagnoza se donosi kad se ustanovi da su pamćenje, vidno-prostorne sposobnosti, mišljenje i ponašanje relativno neoštećeni, a jezični deficit jedini kompromitira svakodnevne aktivnosti kroz najmanje dvije godine trajanja bolesti. Iako jezični poremećaj u PPA može remetiti sposobnost verbalnog pamćenja, bolesnici općenito nemaju problema ni s epizodičkim niti semantičkim pamćenjem. Većina bolesnika nema poremetnji u drugim kognitivnim domenama obično kroz 5-10 godina, a opisani su i slučajevi izolirane poremetnje jezičnih funkcija u trajanju duljem od 15 godina. Bolest obično nastupa prije 65. godine starosti i nešto češće obolijevaju muškarci. Najčešći patološki proces (u oko 60% bolesnika) povezan s PPA je žarišna degeneracija karakterizirana nespecifičnim gubitkom neurona i gliozom i blagim spongioformnim promjenama unutar površnih kortikalnih slojeva. U oko 20% bolesnika je uzrok Pickova bolest s Pickovim tjelešcima (Pickova bolest u užem smislu). I bolesnici s nasljednim tauopatijama, te neki oblici Creutzfeldt-Jacobove bolesti mogu razviti kliničku sliku PPA, ali su tada obično prisutni i dodatni kognitivni i motorički deficiti. Za razliku od afazije u AD, koja je gotovo uvijek tečna (stražnja ili Wernickeova afazija), afazija u PPA je u većine bolesnika netečna (prednja ili Broca afazija). Primarna oštećenja izvan netečne afazije u PPA ponekad uključuju akalkuliju, ideomotornu apraksiju i čistu gluhoću za riječi. Ako pored PPA dođe i do poremećaja razumijevanja, nastaje tzv. ‘semantička demencija’.
Za otprilike 50% slučajeva FTD s parkinsonizmom (FTDP-17, od ‘frontotemporal dementia with parkinsonism’) je 1996. godine otkriveno da ih u 90% bolesnika uzrokuju autosomno dominantne mutacije tau gena na kromosomu 17. Četiri mutacije introna 10 (na pozicijama 3., 13., 14. i 16. kodona) tau gena izazivaju povećano sparivanja eksona 10 i posljedično veće proporcije tau izo-oblika s 4 (a ne 3R) vezna mjesta za MT (izo-oblici 1, 3 i 5). Ovaj poremećaj dovodi do povećanja nevezanog tau i njegovoj agregiranja u filamente (tzv. mutacije s viškom funkcije). Ovi se filamenti mogu naći u neuronima i u gliji, široki su 6-22 nm i svijaju se svakih 90-300nm. Mutacije eksona 10 (L284L, P301L, P301S, S305N) također dovode do FTDP-17 s nakupljanjem tau filamenata, jer mutirani tau (iako s 4R) ima smanjenu sposobnost vezanja za MT (tzv. mutacije s manjkom funkcije), dok mutacije eksona 9 (K257T, L260V, G272V), 12 (V337M) i 13 (G389R, R406W) daju normalan omjer 4R/3R, a u ovim slučajevima se ne oblikuju opisani tau filamenti već ravni filamenti (SF) i PHF. Jedini klasični neuropatološki kriterij koji omogućuje razlikovanje FTDP-17 od FTD su obilni filamentozni depoziti tau proteina u neuronima, a katkad i u gliji. Osim tipa Seattle A, većina bolesnika iz obitelji s FTDP-17, kao i bolesnici s progresivnom supranuklearnom kljenuti i kortikobazalnom degeneracijom, pokazuju elektroforetski profil tau proteina koji se sastoji od dvije pruge od 64 i 68 kDa.
Do sredine 90-tih godina, dijagnoza Pickove bolesti koristila se u širem smislu za bilo koju lobarnu ili žarišnu atrofiju. No, u užem smislu, koji se danas preporučuje, dijagnoza Pickove bolesti može se donijeti samo kad se identificiraju Pickova tjelešca. Ovakva je Pickova bolest relativno rijetka i vidi se u manje od 2% bolesnika s demencijom. Većina slučajeva je sporadična, ali u oko 20% slučajeva vidi se autosomno dominantno naslijeđivanje. Dob početka bolesti varira od 45-65 godina. Početak je podmukao, a progresija može biti brža nego u AD, obično dovodeći do smrti u rasponu od 5-10 godina. Pickova tjelešca su intraneuronalne okruglaste, argirofilne (ali Gallyas-negativne), ubikvitin-pozitivne nakupine koje sadrže fosforilirane neurofilamente i tau proteine. U mnogim slučajevima vide se i balonirani, hipokromatični ‘Pickovi neuroni’ što sadrže fosforilirane neurofilamente i imunoreaktivni su na aB-kristalin. Parijetalna i okcipitalna područja obično nisu zahvaćena, ali su opisane i panencefalitičke i parijetalne varijante. Hipokampalna formacija često pokazuje izraženu atrofiju povezanu s visokom gustoćom Pickovih tjelešaca, naročito u girusu dentatusu. Od subkortikalnih struktura, patološke promjene se najčešće vide u bazalnim ganglijima, amigdalama, bazalnoj i crnoj jezgri, lokusu ceruleusu i rafe jezgrama. Područja koja sadrže Pickova tjelešca i Pickove neurone pokazuju izraziti gubitak neurona i gliozu. Raspodjela Pickovih tjelešaca u korteksu kao i u većini drugih žarišnih atrofija (ali za razliku od raspodjele NFT u AD) često je asimetrična i zahvaća prvenstveno male piramidne neurone II i površnog III sloja, te ponekad i neurone sloja VI. Ultrastrukturalno se Pickova tjelešca sastoje od razbacanih snopova 15 nm širokih ravnih filamenata, koji mogu biti pomiješani s PHF. Pickova tjelešca su imunoreaktivna na fosforilirane epitope neurofilamenata identične onima što se nađu u NFT, te na tau i ubikvitin, što pokazuje da, kao i NFT, možda nastaju iz promijenjenih dijelova normalnog citoskeleta. Štoviše, u većine bolesnika s Pickovom bolešću mogu se istovremeno naći i Pickova tjelešca i NFT, dok se depoziti Ab nađu u oko 30% slučajeva. Ovo ukazuje da, iako imaju različitu laminarnu raspodjelu patoloških promjena, AD i Pickova bolest dijele neke zajedničke biokemijske mehanizme što dovode do sličnih promjena na razini stanice. Kolinergička inervacija korteksa uglavnom je sačuvana. Raspodjela patoloških promjena u Pickovoj bolesti varira od bolesnika do bolesnika, ali uglavnom najviše zahvaća prefrontalni i temporopolarni korteks. U nekim slučajevima poremećaji ponašanja mogu biti dramatični, a uključuju hiperseksualnost, hiperoralnost i disinhibiciju. U zavisnosti od anatomske lokacije patoloških promjena, Pickova bolest može jednako tako izazvati kliničku sliku PPA, progresivne amnezije ili sindroma čeonog režnja. Pored patologije neurona u Pickovoj bolesti je izražena i tau patologija glija stanica. Tau- i ubikvitin-imunoreaktivne nakupine vide se u astrocitima u korteksu i bijeloj tvari. Pored ovog obilježja, činjenice da tau proteini u Pickovoj bolesti sadrže samo izo-oblike s 3R, a elektroforetski obrazac tau proteina pruge na 55 i 64 kDa, osiguravaju pouzdano prepoznavanje Pickove bolesti u odnosu na druge tauopatije.
Frontotemporalna demencija s bolešću motoneurona (FTD-MND, od 'frontotemporal dementia with motor neuron disease') razlikuje se od tipične FTD jer patološke promjene pretežno zahvaćaju hipokampus, jezgru hipoglosusa i motoneurone prednjih rogova medule spinalis. Povezanost između FTD i bolesti motoneurona prvi je opisao Meyer 1929. godine. Početak bolesti varira između 30 i 66 godina starosti, s prosjekom od oko 55 godina, bolest traje 1-6 godina, u prosjeku 2-3, a smrt nastupa uslijed denervacije respiratornih mišića. Muškarci obolijevaju nešto češće od žena. Tipičan je gubitak neurona crne jezgre motoneurona prednjih rogova leđne moždine. Tau proteini u FTD-MND daju tri elektroforetske pruge od 55, 64 i 69 kDa. Imunocitokemijski je bolest u većine slučajeva karakterizirana prisutnošću ubikvitin-pozitivnih, a tau-negativnih intraneuronalnih nakupina (što se ne može vidjeti klasičnim histološkim bojanjima), koje se najčešće nađu u girusu dentatusu i čeonom korteksu. Što je sadržaj ovih nakupina još nije poznato, no slične se nakupine mogu vidjeti i u amiotrofičnoj lateralnoj sklerozi i aksonalnoj distrofiji, a neki obiteljski slučajevi FTD-MND povezani su s mutacijom superoksid dismutaze na 21. kromosomu. Stoga su potrebna daljnja istraživanja da bi se s većom sigurnošću razdvojile ove bolesti.
Uslijed otkrića mnogobrojnih nasljednih tau mutacija koje ubrzavaju agregaciju tau proteina u filamente što su se zbila u posljednje 4 godine, 'rodila se' cijela nova skupina neurodegenerativnih bolesti s demencijom kao glavnim simptomom - nasljedne tauopatije. Pored navedenih nasljednih tauopatija, u ovu skupinu bolesti spadaju još i nasljedna disfazička disinhibicijska demencija (HDDD, od ‘hereditary dysphasic disinhibition dementia’), disinhibicijska demencija s parkinsonizmom i amiotrofijom (DDPAC, od ‘disinhibition dementia with parkinsonism and amyotrophy complex’), palidopontonigralna degeneracija, progresivna subkortikalna glioza, obiteljska višesistemska tauopatija, te sindrom ponašanja sličnog schizophreniji s degeneracijom amigdala (SBAD, od ‘schizophrenia-like behavior with amygdala degeneration’). U novije vrijeme otkrivena je u Danskoj i povezanost FTD s genom na kromosomu 3. Općenito govoreći, motorička su oštećenja u nasljednim tauopatijama jače izražena od onih u sporadičnim žarišnim (lobarnim) atrofijama.
3.3. Bolest Lewyjevih tjelešaca
Bolesti u kojima se pojavljuju Lewyjeva tjelešca raznolike su, uzrok im nije poznat, a zajednička su im neuropatološka obilježja, pored stvaranja Lewyjevih tjelešaca, odumiranje populacija neurona unutar nigralnog dopaminergičkog sustava i medijalnog sljepoočnog područja, te opsežna degeneracija neurita i spongioformna vakuolizacija. Posljednja klasifikacija obuhvaća najmanje 18 različitih oblika bolesti Lewyjevih tjelešaca (LBD, od 'Lewy-body disease') karakteriziranih demencijom i različitim stupnjem parkinsonizma, tako da su ove bolesti vjerojatno sveukupno drugi najčešći uzrok demencije (s učestalošću od oko 20%). Lewyjeva tjelešca su male intracitoplazmatske, eozinofilne, ubikvitin-pozitivne nakupine u neuronima. Gusto eozinofilno središte okruženo je slabije obojanom periferijom (halo). Lewyjeva tjelešca sačinjena su od fosforiliranih neurofilamenata i a–sinukleina. Abnormalna agregacija a-sinukleina vidi se također i u distrofičnim neuritima. Lewyjeva tjelešca mogu se vidjeti u crnoj jezgri bolesnika s idiopatskom Parkinsonovom bolešću s ili bez demencije. Neokortikalna Lewyjeva tjelešca u neuronima male i srednje veličine u V i VI sloju korteksa nisu tako izrazita kao ona u crnoj jezgri i nemaju koncentričnu laminarnu strukturu, a najčešće se nađu u cingularnom i parahipokampalnom girusu, inzuli, sljepoočnom korteksu i amigdalama, te ponekad u čeonom korteksu i hipokampusu. Ako se nađu u korteksu, onda ih gotovo sigurno ima i u bazalnoj jezgri, lokusu ceruleusu, rafe jezgrama i hipotalamusu.
Mehanizmi kojima poremećena funkcija ili agregacija a-sinukleina dovodi do odumiranja neurona nije jasna, ali se čini da su to prvenstveno oštećenje sinapsi i disfunkcija mitohondrija. a-sinuklein je otkriven 1988. godine, a naziv mu dolazi od činjenice da jer je izoliran iz presinaptičkih završetaka i nukleusa. Iz neurona s granulovakuolarnom degeneracijom a- i b-sinuklein izolirani su 1994. godine, a 1995. i njihov homolog u ptica synelfin kritičan za razvojnu plastičnost. Ipak, prekretnica u istraživanjima bila je 1997. godine otkrivena mutacija A53T gena za a-sinuklein na 4. kromosomu u talijanskih i grčkih obitelji s autosomno-dominantnim naslijeđivanjem Parkinsonove bolesti (te 1998. godine mutacija A30P u njemačkih obitelji s Parkinsonovom bolešću). Danas znamo da mutacije koje mijenjaju konformaciju a-sinukleina uzrokuju nasljedne oblike LBD.
Ipak, tipična LBD je poremećaj povezan s poodmaklom životnom dobi, a obiteljsko pojavljivanje je rijetko. Simptomi mogu biti povezani s bilo kojim dijelom moždane kore, no obično je glavno obilježje bolesti fluktuirajuće stanje konfuzije s vidnim iluzijama i halucinacijama. Težina demencije proporcionalna je gustoći Lewyjevih tjelešaca. U kliničkoj slici također su karakteristične halucinacije i u drugim modalitetima, te prolazni gubici svijesti. Za razliku od AD, difuzna bolest Lewyjevih tjelešaca ima relativno rane ekstrapiramidne simptome što nastupaju nakon 4-5 godina trajanja bolesti, kognitivne smetnje karakteristično osciliraju, vidne halucinacije i depresija česte su već u ranim stadijima bolesti, a hipokampus i pamćenje obično su dugo sačuvani. Zanimljivo je također da i do 50% ovih bolesnika ima normotenzivni hidrocefalus. U mnogih bolesnika s LBD, postmortalno se vidi i spongioformna vakuolizacija što podsjeća na Creutzfeldt-Jacobovu bolest, no u LBD je ona ograničena na entorinalni i sljepoočni korteks, amigdala i katkad cingularni girus. Čini se da vakuole u ovom slučaju nastaju uslijed degeneracije terminalnih aksona velikih neurona IIIC i V sloja piramidnih neurona transentorinalnog korteksa. Kolinergička kortikalna inervacija je karakteristično jače oštećena nego u AD. Potvrda za navedeni nalaz je i činjenica da većina kolinomimetika bolje djeluje na kognitivnih status bolesnika s LBD nego AD.
Ponekad se Lewyjeva tjelešca vide u dementnih bolesnika s AD, pa se postavlja dijagnoza AD s DLBD (od ‘diffuse Lewy-body disease'). U drugih se pak dementnih bolesnika mogu vidjeti Lewyjeva tjelešca u prisutnosti SP, ali bez NFT što se naziva Lewyjevom varijantom AD (od 'Lewy body variant of AD'). U trećoj skupini dementnih bolesnika se vide LB bez bilo kakvih drugih histopatoloških promjena ili fokalne atrofije. Za ovu je skupinu zasad najprikladniji naziv difuzna LBD (DLBD, od 'diffuse LBD').
Parkinsonizam u LBD slabo reagira na dopaminergičke lijekove. Bolesnici s DLBD također su neobično osjetljivi na tipične i atipične neuroleptike. Ovi lijekovi u njih vrlo često izazivaju nuspojave, a također mogu dovesti do pogoršanja demencije i neuroleptičkog malignog sindroma.
Pored povezanosti s nastankom Lewyjevih tjelešcaca u LBD, abnormalna agregacija i akumulacija a-sinukleina može biti povezana i s nastankom SP u AD. Naime, 1993. godine je otkriveno da je a-sinuklein prekursor fragmenta koji se nalazi u neamiloidnom dijelu SP (NACP, od ‘non-Ab component precursor of AD amyloid’).
3.4. Bolest argirofilnih zrnaca (Braakova demencija)
Argirofilna Braakova zrnca (BAG, od 'Braak's argyrophilic grains') su prvi opisali Braak i Braak 1987. godine kao neuropatološko obilježje u dementnih osoba, no pokazalo se da se ove promjene mogu naći u manjem broju i u neuropilu moždanog tkiva normalnih starijih osoba. BAG su male, vretenaste argirofilne strukture, a dendriti koji ih sadrže progresivno se skvrčavaju. Za razliku od NT koji imaju oblik niti, BAG imaju oblik banane. BAG predstavljaju još jedan morfološki oblik patološke hiperfosforilacije tau proteina. Pa ipak, za razliku od klasične neuropatološke slike u AD, iako se u BAG odvija hiperfosforilacija tau proteina, često ne dolazi do nastanka NFT, čak niti u slučajevima s ogromnim brojem BAG. Ako su BAG jedini patološki nalaz u dementne osobe, predloženo je da se takav nozološki entitet naziva bolest argirofilnih zrnaca (AGD, od 'argyrophilic grain disease' ili Braakova demencija). U slučajnom postmortalnom uzorku od 2661 mozga u 6% je neuropatološki nađena AD, a u 5% Braakova bolest. Nalaz BAG ipak nije specifičan, jer se ova zrnca mogu susresti i u drugim bolestima i stanjima, npr. Pickovoj bolesti, kortikobazalnoj degeneraciji, a naročito su česta u progresivnoj supranuklearnoj kljenuti. Pored BAG često se javljaju okruglasta tjelešca ('coiled bodies') u bijeloj tvari, a ona se jednako kao i BAG, elektronsko-mikroskopski sastoje od ravnih tubula promjera 25 nm. Na temelju morfološke sličnosti, čini se da okruglasta tjelešca predstavljaju degenerirane oligodendrocite. Bolest argirofilnih zrnaca spada u tzv. 4R tauopatiju (vidi shemu 2). S obzirom na činjenice da se relativno visoka učestalost argirofilnih zrnaca viđa u PSP (u oko 20% oboljelih) i CBD (u oko 40% oboljelih), ta da argirofilna zrnca u PSP i CBD nisu ograničena na limbički režanj i hipotalamus, moglo bi se zaključiti da 4R tauopatije predstavljaju klinički spektar bolesti. Tako bi Braakova demencija predstavljala 4R tauopatiju medijalnog dijela temporalnog režnja i nalazila se između difuzne kortikalno-subkortikalne tauopatije kakva se vidi u CBD i više ograničene tauopatije bazalnih ganglija i moždanog debla kakva se vidi u PSP.
Nakon akutne alkoholne krize bolesnik može pasti u stupor ili komu, nastaju nistagmus i paraliza okulomotornih mišića, vide se nejednake zjenice, ataksija, periferne neuropatije i sindrom diencefaličke amnezije (Wernickeova encefalopatija). U dijela bolesnika koji se uspiju oporaviti zaostaje teška amnezija i smetnje koncentracije, obično u odsutnosti simptoma čeonog režnja (Korsakoffljeva amnezija ili Wernicke-Korsakoffljev sindrom). Prestankom uzimanja alkohola simptomi amnezije mogu se značajno smanjiti.
U kroničnih alkoholičara s progresivnom demencijom vide se pored amnezije i znakovi disfunkcije čeonog režnja, a oporavak je značajno slabiji. Neuropatološke promjene povezane s kroničnim alkoholizmom nastaju uglavnom kao posljedica nedostatka tiamina (vitamin B1), ali samo u slučaju njegovog slabijeg unosa hranom i genetske predispozicije ulijed slabije sposobnosti vezanja transketolaze za tiamin-pirofosfat (što objašnjava zašto samo mali broj kroničnih alkoholičara oboli od Wernicke-Korsakoffljevog sindroma).
Glavna neuropatološka oštećenja u Wernickeovoj encefalopatiji nalaze se u mamilarnim tijelima i područjima oko 3. i 4. moždane komore i akvedukta. Makroskopski se često mogu uočiti petehijalna krvarenja, obično u mamilarnim tijelima, ali i u međumozgu i moždanom deblu. Mikroskopski se vide dilatacija kapilara i krvarenja. U Wernicke-Korsakoffljevom sindromu mamilarna tijela su skvrčena, te pokazuju značajan gubitak neurona i reaktivnu astrocitozu, što se često vidi i u medijalnom dijelu talamusa.
Prionske bolesti čovjeka uključuju Creutzfeldt-Jacobovu bolest (CJD), Gerstmann-Sträussler-Sheinkerov sindrom (GSS), smrtonosnu obiteljsku nesanicu (FFI, od ‘fatal familial insomnia’) i kuru. Sve su ove bolesti karakterizirane akumulacijom abnormalnog, zbog promjenjene kiralnosti nekih aminokiselina na proteaze i toplinu super-otpornog, prionskog glikoproteina (PrP, od ‘prion protein’) u moždanoj kori i drugim dijelovima živčanog sustava.
Prionske bolesti mogu nastati na tri načina: horizontalnom transmisijom, nasljednim mutacijama gena prionskog proteina (PrP, od ‘prion protein’) i sponatanom mutacijom. Horizontalni prijenos bolesti s ovce na govedo (BSE, od ‘bovine spongioform encephalopathy’), počeo se događati u masovnim razmjerima 1985. godine u Velikoj Britaniji. U goveda tovljenih smrvljenim ostacima ovaca zaraženih scrapijem, patološki, infektivni ovčji prionski protein, PrPSc, iz probavne cijevi dospjeva u mozak goveda gdje se akumulira i vremenom pretvara normalni prionski protein, PrPc , u PrPSc. Zbog dugogodišnjeg zataškavanja ovog nalaza od strane Vlade Velike Britanije, početkom 90-tih godina broj zaraženih goveda narastao je na preko 40000. Jedenjem zaraženih goveda, bolest je prešla na čovjeka (prvi slučaj opisan je 1996. godine), a nazvana je novom varijantom CJD (vCJD, od ‘variant CJD’) od koje je do konca 2001. godine u Velikoj Britaniji umrlo više od 100 ljudi (jedna je majka bolest transplacentarno prenijela i na dijete).
Kuru je opisan u Foré plemena što obitava u istočnim visoravnima Papua Nove Gvineje, a prenosi se ritualističkim kanibalizmom. Zbog zabrane jedenja ljudi broj je oboljelih u novije vrijeme drastično smanjen. Ataksija je glavni simptom kurua (kuru na jeziku ovog plemena znači ‘tresti se’). Bolest je vjerojatno nastala iz inicijalnog sporadičnog slučaja CJD, a dalje se prenosila jedenjem zaraženog moždanog tkiva.
CJD je opisana početkom 20-tih godina XX stoljeća. Javlja se s prevalencijom od oko 1:2 milijuna stanovnika godišnje, no ima područja gdje je učestalost i značajno veća. Najčešći su simptomi demencija i mioklonus. Smrt uslijedi obično nakon 3-12 mjeseci od početka bolesti. Bolest je uglavnom sporadična, ali u 10-15% slučajeva naslijeđuje se autosomno dominantno, što je razlogom geografskih razlika u učestalosti. FFI je karakterizirana neizlječivom nesanicom i disautonomijom, a neuropatološki se vidi selektivna degeneracija pojedinih dijelova talamusa, pa se naziva i ‘talamičkim tipom CJD’. GSS je opisan između 1928. i 1936. godine, a incidencija mu je 1-10:100 milijuna stanovnika. U ovih bolesnika ataksija obično nastupi prije demencije. Progredira sporije od CJD, tako da traje u prosjeku dulje od 5 godina. Obično se naslijeđuje autosomno dominantno.
Dok su GSS i FFI su pretežno nasljedni sindromi povezani s mutacijama PrP gena, CJD uključuje sporadične, obiteljske i prijenosne oblike bolesti. CJD se najčešće prenosi jatrogeno putem nesteriliziranih instrumenata pri biopsijama, krvnim produktima, te transplantacijom dure mater i korneje. Između 1963. i 1985. godine zaražen je veliki broj djece malog rasta i neplodnih žena davanjem hormona rasta, odnosno gonadotropina izoliranih iz hipofiza kadavera (zbog duge inkubacije, poremećaj je dugo vremena ostao neprepoznat).
U većine bolesnika s GSS (te u 15% CJD slučajeva i većine obduciranih bolesnika s kuruom), vidi se jasno izraženo izvanstanično odlaganje prionskog amiloidnog proteina u obliku ‘prionskih plakova’. Prionski protein je u čovjeka kodiran genom na kratkom kraku kromosoma 20, sadrži 253 kodona unutar istog eksona, najviše se eksprimira u živčanim stanicama, a funkcija mu nije poznata (u miševa s homozigotnom delecijom nisu uočene promjene).
Heterogenost mutacija PrP gena smatra se ponajviše odgovornom za različitost kliničkih obilježja prionskih bolesti. Štoviše, iako obiteljska CJD i FFI mogu biti povezane s istom mutacijom kodona 178 PrP gena (asparagin umjesto aspartata), polimorfizam kodona 129 određuje hoće li nastati CJD (valin) ili FFI (metionin). Polimorfizmi kodona 129 također su povezani s različitim kliničkim oblicima sporadične CJD, kao što su mioklona, Heidenhainova i ataksijska varijanta, a određuju i prijemčljivost za HSE. Naime, koliko je dosad poznato, svi oboljeli od vCJD su homozigoti za metionin na 129 kodonu PrP gena (inače je među bijelom rasom oko 43% homozigota za metionin na kodonu 129). Kako većina prionskih bolesti dovodi pored demencije i do kliničkih obilježja povezanih s motoričkim sustavom, npr. mioklonusa i ataksije u CJD ili cerebelo-piramidnog sindroma u GSS, ovaj nalaz olakšava njihovo rano razlikovanje u odnosu na druge primarne demencije.
Čini se da neuropatologija vCJD pokazuje neka obilježja drugačija od drugih prionskih bolesti: tzv. floridne prionske plakove u moždanoj kori i malom mozgu, spongioformna promjena najčešća je u kaudatusu i putamenu, gubitak neurona i glioza najjače su izraženi u stražnjem talamusu, a akumulacija abnormalnog izo-oblika prionskog proteina izražena je i u limfoidnim tkivima.
4. ‘Normalno’ starenje i demencija
Mnogobrojna su istraživanja potvrdila prisutnost i NFT i SP u moždanoj kori nedementnih starijih osoba. Predilekcijska mjesta za nastanak ovih promjena vrlo su slična kao i u AD: površni slojevi entorinalne moždane kore, CA1 područje, prosubikulum i amigdala. Odlaganje amiloida i nastanak NFT u nekih su normalnih, nedementnih osoba prisutni su u tolikoj mjeri da se mogu mjeriti čak i sa vrijednostima dementnih osoba s AD. Tako su opisani slučajevi sa visokim gustoćama NFT i SP u hipokampalnoj formaciji (i eventualno sljepoočnoj moždanoj kori), a rijetkim NFT u ostalim područjima neokorteksa, koji su imali samo vrlo blage kognitivne poremećaje. Ovakve osobe obično se razlikuju od bolesnika s AD po tome što im se u DN plakova ne nalaze PHF, što znači da imaju puno manji broj NP.
Proučavanja stogodišnjaka pokazala su da jedan vrlo mali postotak ove populacije ima vrlo mali broj NFT samo u II sloju entorinalnog korteksa, te ponekad još i u CA1 području, subikulumu i donjoj sljepoočnoj moždanoj kori (BA 20). Neki istraživači pretpostavljaju da ova skupina ljudi predstavlja posebnu genetsku skupinu, a ovakav nalaz se u neuropatologiji katkad naziva i 'čisto starenje'. Zbog toga ima i prijedloga da se svi ljudi koji imaju NFT i SP trebaju svrstati u klinički nezamjetnu, a neuropatološki blagu AD.
Demencija je jedan od najčešćih sindroma što se susreću u svakodnevnoj praksi neurologa, neuropsihijatra i neuropsihologa. Uslijed raznolikosti uzroka, višebrojnosti žarišta i postepene progresije, sindrom demencije je i klinički i neuropatološki vrlo heterogen. Poremećaje ponašanja koji nastaju kao posljedica demencije treba promatrati kroz načela anatomsko-funkcionalne korelacije zahvaćenih dijelova mozga i neuropsihološkog statusa bolesnika. Pri tome treba imati na umu da kortikalna područja pogođena poremećajima što dovode do demencije gotovo nikad nisu u potpunosti uništena, već postoji različit stupanj selektivnosti s obzirom na tipove neurona i njihov anatomski smještaj. Učinci višestrukih oštećenja nisu samo aditivni, već uključuju složene interakcije, a polagana progresija također omogućuje veći stupanj funkcionalne reorganizacije no što je uobičajeno nakon akutnih oštećenja. Ne postoji “tipična demencija”, već svakog dementnog bolesnika treba promatrati individualno i u zavisnosti od osobnosti prije nastanka demencije.
Neurološki i neuropsihološki deficiti u različitim dementnim stanjima više odražavaju anatomsku raspodjelu patoloških promjena nego narav samog uzroka. Tako npr. Alzheimerova i Pickova bolest mogu imati istu kliničku sliku ako su neurofibrilarni snopići i Pickova tjelešca anatomski slično raspodijeljeni, ali klinička slika može biti i vrlo različita ako je raspodjela navedenih promjena drugačija. Svaki poremećaj ili bolest koji dovodi do sindroma demencije ipak pokazuje određeni obrazac predilekcijskih mjesta, tako da se iz simptomatologije obično može naslutiti narav uzroka (npr. neurofibrilarni snopići u Alzheimerovoj bolesti imaju predilekciju za limbički, a Pickova bolest za čeoni i temporopolarni korteks).
Daleko najčešći uzrok demencije je Alzheimerova bolest. Genetska istraživanja
ukazuju na beta-amiloid kao ključni patogenetski činitelj u nastanku Alzheimerove
bolesti, dok kliničko-patološka korelacija upućuje na veću važnost patologije
tau proteina i neurofibrilarne degeneracije. Hipoteza sloma neuronalne plastičnosti
zasad jedina primjereno i sveobuhvatno povezuje sva genetska (mutacije prekursornog
proteina za amiloid, presenilina 1 i 2, te polimorfizam apolipoproteina E) i
neuropatološka obilježja bolesti (neuritički plakovi i neurofibrilarni snopići),
te druge poznate rizične činitelje (dob, trauma glave) što utječu na nastanak
Alzheimerove bolesti. Hipoteza se temelji na zapažanju da su izrastanje aksonskih
mladica, dugoročna potencijacija sinapsi, preoblikovanje dendrita i reaktivna
sinaptogeneza najjače izraženi u limbičkim i bazalnim područjima mozga, koja
uslijed nemogućnosti neprekidnog ispunjavanja zahtjeva za plastičnošću nakon
30 godina života prva pokazuju patološke promjene. Kao odgovor na ovakav stres
dolazi do povećane sinteze prekursornog proteina za amiloid i stvaranja veće
količine beta-amiloida. Među brojnim istraživačima danas prevladava mišljenje
da je fibrilogeneza difuznih depozita beta-amiloida vjerojatno jedan od glavnih
mehanizama koji dovodi do fizičkog oštećenja neurona uslijed kompromitiranja
aksonskog transporta. Odgovor neurona na ovakvu vrst oštećenja rezultira mobilizacijom
i prekomjernom fosforilacijom tau proteina. Hiperfosforilacija dovodi do polimerizacije
tau proteina nevezanih za mikrotubule i stvaranja ravnih i sparenih uzvojitih
filamenata čijim nakupljanjem nastaju distrofički neuriti senilnih plakova,
neurofibrilarni snopići i neuropilne niti. Ove neuropatološke promjene karakteriziraju
Alzheimerovu bolest, a u manjoj mjeri se susreću i u mnogim drugim neurodegenerativnim
bolestima i ‘normalnom’ starenju.